光分解離子成像
光分解離子成像,或更普遍地來說,產物成像是一種測量化學反應或光分解產物速度分佈的實驗技術 [1]。 該方法使用二維偵測器,通常是微通道板,來擷取透過共振增強多光子離子化之態選擇後的離子到達偵測器的位置。第一個光分解離子成像實驗是由大衛·錢德勒(David W. Chandler)和保羅·休斯頓(Paul L. Houston)在1987年完成,其題目為碘甲烷的光分解動態學[2]。
背景
許多分子反應動態學的問題需要同時測量粒子的移動能和角分佈,亦或是速度分佈;甚至還需要測量與該粒子速度相依的內能。研究分子反應,能量轉移過程和光分解等,只有在所有產物的內能和速度都清楚的情況下才能全盤了解。產物成像方法可以透過測量某一經過態選擇後的產物之三維速度分佈來達成此一目標。對於一個產生兩種產物的反應,如光分解反應 ABC(i) + hν → A(f1) + BC(f2) 或分子反應 AB(i1) + C(i2) → A(f1) + BC(f2),透過選擇性地測量 A 產物之 f1 態的速度分佈,我們通常可以用動量守恆與能量守恆的關係來推斷沒有被測量的產物 BC 之內能(或是 f2 態,或是能階)分佈。
範例
底下用一個簡單的例子來說明此原理。臭氧(O3)經過紫外光(UV)激發後,產生氧原子和氧分子,主產物為 O(1D) 和 O2(1Δ),也就是說,氧原子和氧分子都是在他們的第一電子激發態。在光波長為 266 nm 時,有足夠的能量來解離臭氧而生成此二產物,並激發 O2(1Δ) 最高到 v = 3 的振動態,並提供一些能量來彈開兩個分解產物,轉變為其速度。其反應式如下:
O3 + hν266 nm → O(1D) + O2(1Δ, v = 0-3, j) + 移動能
想當然爾,越多的的能量用在激發 O2 的振動(v)或轉動(j),彈開的產物就只能獲得越少的移動能,速率也就越低。用REMPI來選擇性地離子化 O(1D) 原子,並配合使用產物成像技術,得到的映像,可以用來判定 O(1D) 三維的速度分佈,並從而推得 O2(1Δ) 的內能分佈。如右圖[3],圓圈由外而內分別代表 O2(1Δ) 的振動態由 v = 0 - 3,而每個圈由外緣往內亦代表轉動態 j 由小而大的分佈。此外,產物的角分佈也是用來判定反應通道與產物極化 (polarization) 的重要資料。
產物成像術
在最初的產物成像論文中,是離子的位置與速度同時被映像到二維的偵測器。首先是一道雷射光解離碘甲烷(CH3I 分子),緊接著另一道雷射光運用REMPI來選擇性離子化特定振動態的甲基(CH3)產物。這兩道雷射光都是脈衝式的,而且離子化雷射光相對於光分解雷射光的延遲要夠短,才能在產物還沒飛離時把它離子化。由於電子的質量遠小於甲基,在離子化過程中彈射出的電子幾乎不會改變彈射出的甲基離子之速度,所以我們大致可以忽略其影響。當甲基產物被離子化後,即被平行板電場加速,將離子投射到二維偵測器。此偵測器通常是由兩片微通道板 (Chevron-MCP)、磷光屏 (PS) 及CCD相機組成。
產物成像術的改進
速度映像 (Velocity Map Imaging)
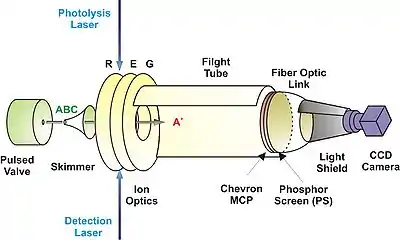
安德烈•埃品客 (André T. J. B. Eppink) 和大衛•派克 (David H. Parker) 在十年後 (1997年) 大幅度地改善了產物成像術 [4]。 基本上,其設計使用的加速電場設計與威利 (Wiley) 和邁凱倫 (McLaren) [5] 的飛行時間質譜儀很類似,透過三片平行電極板(Repeller, Extractor和Ground)來加速離子到二維偵測器。埃品客和派克巧妙地移除在Extractor和Ground電極上的Grid而達到離子透鏡聚焦的效果。在適當的離子透鏡電位設定下,離子到達偵測器的位置只與離子的初始速度有關,而與初始位置無關。此一舉突破性地增進速度解析度,而在分子反應動態學界掀起了產物速度映像的研究熱潮。
速度映像,基本上就是離子映射到偵測器的位置 x 正比於其初始速度 vx。由於 x 正比於 vx • TOF (飛行時間),而 TOF 正比於 ,所以我們可以了解,x 正比於離子在 x 方向的初始能量 Ux 開根號,而與粒子的質量無關。這使得電子成像 (Electron imaging) 也成為可能。此外,由於原始的速度映像是把 vx, vy 和 vz 投射到偵測器的 x 和 y,使得三維的訊息被投射到二維映像。通常這個映像會有一個圓柱對稱軸,可以透過 Abel transform 來轉換成純粹 vx 與 vy 的二維資料。若沒有這個對稱軸,則需要更複雜的轉換程序。因此,一些實驗室也開發純粹二維的映像技術或是三維的映像技術。

二維薄片映像 (2D Slice Imaging)
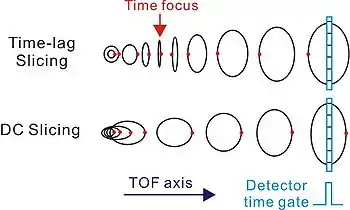
- 時間延遲薄片法 (Time-lag Slicing)
Kitsopoulos實驗室在2001年實作了第一個二維薄片映像,稱為時間延遲薄片法 [6]。 相反與威利-邁凱倫運用一個離子化與電場脈衝的時間延遲,來獲得時間聚焦而增進飛行時間質譜儀的解析度;Kitsopoulos 用這個時間延遲來讓時間失焦,達成二維薄片映像。
- 直流薄片法 (DC Slicing)
在2003年幾乎同時間由劉國平實驗室 [7] 和Suits實驗室 [8] 實作了不需要用此脈衝電場也可以達成二維薄片映像的方法,稱直流薄片法。基本上,直流薄片法是改變電極板的設計,添加更多的電極板,讓離子產生時所處的電場 E1 較為微弱,而讓離子的迴轉時間 (turn-around time) 變長來達成二維薄片成像:

時間延遲薄片法與直流薄片法皆屬於時間薄片成像,需要透過偵測器的時間閘 (time gate) 來選擇切片。派克實驗室也證實了運用傳統三片電極板的設計,在某些條件下也可以用來作時間延遲薄片法與直流薄片法[9]。
- 光學薄片法 (Optical Slicing)
Tonokura 和 Suzuki 在1994年第一次使用光學薄片離子成像法[10]。適當地調整光解雷射與離子化雷射的位置差與脈衝時間差,在離子化時就可以把產物作速度切片,稱為光學薄片法。他們使用兩道雷射薄片 (laser sheet),稱片-片光學薄片法 (sheet-sheet optical slicing)。派克實驗室用點-片幾何 (dot-sheet geometry) 也可以得到很好的光學薄片[9]。另外,Suits 透過點-點幾何 (dot-dot geometry),所得的映像稱光柵成像 (raster imaging)[8]。上面的點-點幾何的兩道雷射是平行的,若由平行改為垂直時,也可以得到完好的光學薄片成像。
- 都卜勒薄片法 (Doppler Slicing)
此外,理論上,也有可能用都卜勒效應來作速度切片,不過此時離子化雷射會直接射向偵測器,所以設計上較為困難,目前尚未有運用都卜勒薄片法的實作。光學薄片法和都卜勒薄片法在離子產生時,就已經選擇性地離子化二維的速度分佈,所以不需要透過偵測器時間閘來切片。
三維離子成像 (3D Ion Imaging)
如果採用三維偵測器,如 delay line anode 或 double exposure CCD camera,可獲得完整三維的映像。也就是說把 vx, vy 和 vz 投射到偵測器的 x 和 y 和 Δt,此方法稱為三維離子成像。
參考文獻
- Whitaker, Benjamin J (ed.), , Cambridge University Press, 2003, ISBN 0-521-810590
- Chandler, David W.; Houston, Paul L., , J. Chem. Phys., 1987, 87: 1445–7, doi:10.1063/1.453276
- Dylewski, S. M.; Geiser, J. D.; Houston, P. L., , J. Chem. Phys., 2001, 115: 7460–7473, doi:10.1063/1.1405439
- Eppink, A. T. J. B.; Parker, D. H., , Rev. Sci. Instrum., 1997, 68: 3477–3484, doi:10.1063/1.1148310
- Wiley, W. C.; McLaren, I. H., , Rev. Sci. Instrum., 1955, 26: 1150–1157, doi:10.1063/1.1715212
- Gebhardt, C. R.; Rakitzis, T. P.; Samartzis, P. C.; Ladopoulos, V.; Kitsopoulos, T. N., , Rev. Sci. Instrum., 2001, 72: 3848–3853, doi:10.1063/1.1403010
- Lin, J. J.; Zhou, J. G.; Shiu, W. C.; Liu, K. P., , Rev. Sci. Instrum., 2003, 74: 2495–2500, doi:10.1063/1.1561604
- Townsend, D.; Minitti, M. P.; Suits, A. G., , Rev. Sci. Instrum., 2003, 74: 2530–2539, doi:10.1063/1.1544053
- Chestakov, D. A.; Wu, S.-M.; Wu, G.; Parker, D. H., , J. Phys. Chem. A, 2004, 108: 8100–8105, doi:10.1021/jp0491111
- Tonokura, K.; Suzuki, T., , Chem. Phys. Lett., 1994, 224: 1–6, doi:10.1016/0009-2614(94)00533-8