巴顿反应
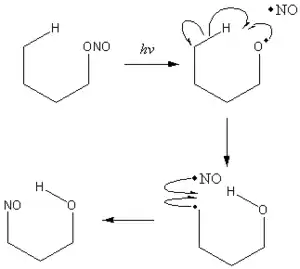
巴顿反应(Barton反应)以英国化学家德里克·巴顿命名,也称为巴顿亚硝酸酯反应[1]反应中亚硝酸酯光解生成δ-亚硝基醇。反应经由RO–NO键均裂,氧自由基夺氢,而后自由基结合的机理。[2]
类似的反应是以卤代胺为原料的Hofmann-Löffler-Freytag反应。
该反应是在1960年发现的,其发现者是诺贝尔奖获得者德里克·巴顿爵士。[3] 巴顿1969年的诺贝尔化学奖是因为他在理解有机分子构象方面的工作而获奖,这项工作对于实现巴顿反应的实用性至关重要。[4]
Barton反应涉及RO-NO均匀断裂,然后进行δ-夺氢反应,自由基重组和互变异构反应形成肟。[5]δ-氢的选择性是6-元基团中间体的构象的结果。 通常,可以容易地预测氢原子夺取的位置。 这允许区域选择性和立体选择性地将功能性引入到具有高产率的复杂分子中。 由于其独特的衍生其他惰性底物的能力,巴顿在20世纪60年代广泛使用这种反应来制造许多非天然的类固醇类似物。 [6]
虽然Barton反应尚未得到许多其他有机反应的普及或广泛使用,同样的是机理上类似的Hofmann-Löffler-Freytag反应,但它代表了碳氢键活化化学的第一个例子,在工业和学术化学界这个领域现在是许多前沿研究的主题。[7]
亚硝酸烷基酯的制备
The unusual 亚硝酸烷基酯starting material of the Barton reaction is prepared by attack of an alcohol on a nitrosylium cation generated in situ by dehydration of doubly protonated nitrous acid.[8] This series of steps is mechanistically identical to the first half of the mechanism formation of the more well-known aryl and alkyl 重氮盐.
While the synthesis of alkyl nitrites from 亚硝酰氯 is known and oft-employed in the context of complex molecule synthesis, the reaction is reversible and the products are in thermodynamic equilibrium with the starting material. Furthermore, nitrosyl chloride is a powerful oxidizing agent, and oxidation of the alcohols with concomitant chlorination has been observed.[9] The reaction of nitrosyl chloride with aromatic alcohols generally yields nitroso compounds and other over-oxidation products.
反应机理和区域选择性
The Barton reaction commences with a photochemically induced cleavage of the nitrite O-N bond, typically using a high pressure mercury lamp.[10] This produces an alkyoxyl radical which immediately abstracts a hydrogen atom from the δ-carbon. In the absence of other radical sources or other proximal reactive groups, the alkyl radical recombines with the nitrosyl radical. The resultant nitroso compounds undergoes 互变异构 to the isolated oxime product.
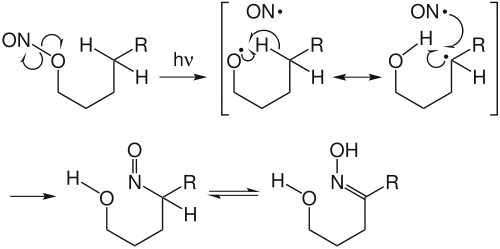
The carbon centered radical can be intercepted by other radical sources such as iodine or 丙烯腈. The first instance results in the δ-hydrogen being replaced with iodine, then subsequent 环化 to a 四氢呋喃 by an SN2反应.[11] The second example results in a chain elongation product with the oxime formed 2 carbon units further from the oxygen than normal.[12]
This mechanistic hypothesis is supported by 动力学同位素效应 experiments.[13] Isotopic labeling of the nitrite with 15N has shown the mechanism non-‘caged’ and that the nitrosyl radical formed from a given nitrite recombines randomly with other alkyl radicals. However, recombination of the nitrosyl radical with the alkoxyl radical (a reversal of the homolytic cleavage) has been shown to proceed without scrambling of isotope labels.[14] This lack of tight radical pairing is also supported by the observation that alkyl radicals generated by Barton conditions can undergo radical cyclization while analogous intermediates generated by 四乙酸铅 oxidation do not.[15]
In rare cases, it appears that the alkoxyl radical may epimerize before hydrogen atom abstraction.[16]
Most commonIy, including steroidal systems, the hydrogen atom is abstracted from a methyl group that has a 1,3 diaxial relationship with the alkoxyl radical.[17] In the absence of a hydrogen on the δ-carbon, or when the particular conformation of the substrate orients the ε-carbon close together, 1,6-hydrogen atom transfer is the favored process. However, these reactions tend to be an order of magnitude slower than the corresponding 1,5-hydrogen atom transfer.
Computational studies have shown that this preference for 1,5-hydrogen atom transfer over 1,6-hydrogen atom transfer appears to be entropically favored rather than a result of a particular stable ‘chair-like’ transition state.[18] In fact, it has been calculated that the 1,6-hydrogen atom transfer proceeds through a transition that is about 0.8 kcal/mol lower than that of the 1,5.
In acyclic systems, δ-hydrogen abstraction is still observed, however, alpha-hydrogen abstraction to form the corresponding ketone competes.[19]
In certain cases, particularly nitrites derived from cyclopentyl alcohols, the oxygen-centered radical prefers to react via C-C bond cleavage as opposed to H-atom abstraction.[11] For example, when subjected to Barton conditions, cyclopentyl nitrite forms glutaraldehyde monoxime. This is also observed in cases where the radical intermediate formed by fragmentation is particularly stable, such as the allylic radical formed by the fragmentation of isopulegol nitrite.[20]
变体
In rigid systems such as aldosterone, the 1,5-hydrogen atom transfer is exceedingly fast, with a rate constant on the order of 10^7 s-1. Similar intermolecular H-atom transfer can be up to 100 times slower.[21] Furthermore, the hydrogen atom transfer benefits from the formation of a stronger O-H bond at the expense of a weaker C-H bond. For the formation of a primary, second, or tertiary alkyl radical from an alkoxyl radical, there is a driving force of 3 kcal/mol, 5 kcal/mol, and 9 kcal/mol, respectively.[17]
The alkyl radical formed after hydrogen atom transfer is susceptible to standard radical reactions when scavengers are present in sufficient excess to outcompete the nitrosyl radical. Soon after their initial disclosure, Barton and co-workers reported the trapping of the radical with I2 and CCl3Br (as Iodine and Bromine radical sources, respectively) to form the δ-halo-alcohol. These halohydrin species can be cyclized to the corresponding 四氢吡喃 derivates under basic conditions.[22]
Large excesses of activated alkenes can be used to intercept the alkyl radical and results in formation of a C-C bond from an unactivated C-H bond.[23]
In the presence of oxygen, the alkyl radical is trapped and forms an organic peroxy radical. This intermiediate is trapped by the nitrosyl radical and then isomerizes to give a δ-nitrate ester which, while both acid- and base-stable, can be reduced to the corresponding alcohol under mild conditions.[24]
在复杂分子合成中的应用
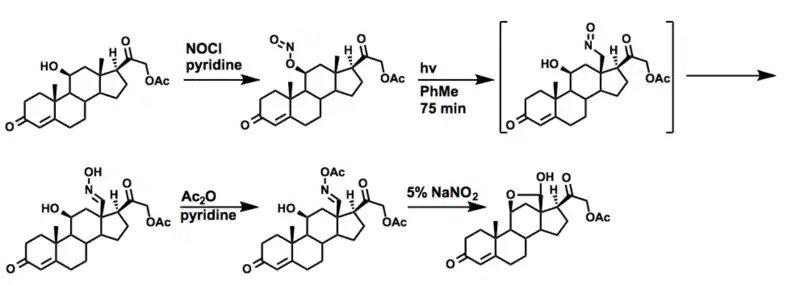
醋酸醛固酮
In a publication immediately proceeding Barton’s initial disclosure of the methodology in the Journal of the American Chemical Society, a synthesis of 醛固酮 acetate is demonstrated.[25] Allowing corticosterone acetate to react with nitrosyl chloride in dry pyridine yields the nitrite. Subsequently, irradiation under inert atmosphere followed by treatment with aqueous sodium nitrite selectively gives the desired oxime. The oxime is then acetylated and hydrolyzed to yield the natural product hemiacetal.
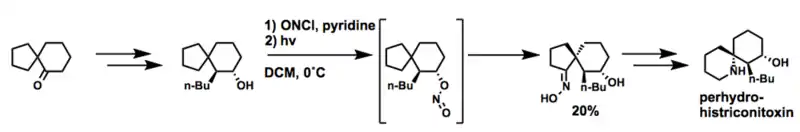
全氢化戏蛙毒素
After a short synthesis to obtain the desired spiro-[5.4] system, Nobel Laureaute E.J. Corey and co-workers employed a Barton reaction to selectively introduce an oxime in a 1,3-diaxial position to the nitrite ester. The oxime is converted to a 内酰胺 via a 贝克曼重排反应 and then reduced to the natural product.[26]
印苦楝二酮
Corey again employed the Barton reaction in the synthesis of Azadiradione, a member of the limonoid family of natural products. In this case, 亚硝酰基硫酸 is used in place of nitrosyl chloride.[27]
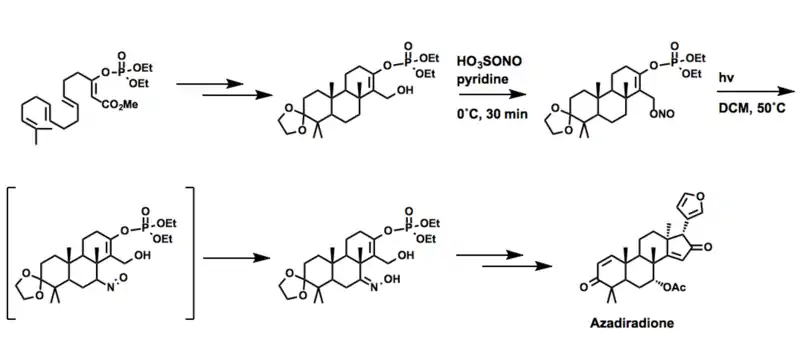
别白桦脂醇衍生物
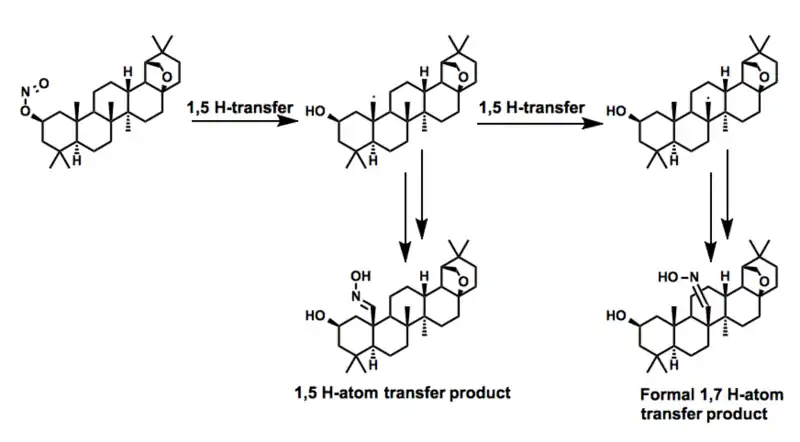
In the process of preparing a series of derivatives of the triterpenoid allobetulin别白桦脂醇, Dehan and coworkers observed a remarkable transformation resulting from two consecutive 1,5-hydrogen atom transfers. While the product of the single 1,5-hydrogen atom transfer was also observed, the former transformation represent a formal 1,7-hydrogen atom transfer across an enormous distance.[28]
参考文献
- D. H. R. Barton, J. M. Beaton, L. E. Geller, and M. M. Pechet. . Journal of the American Chemical Society. 1961, 83 (19): 4076–4083. doi:10.1021/ja01480a030.
- (PDF). [2008-06-27]. (原始内容 (PDF)存档于2007-06-09).
- Barton, D. H. R.; Beaton, J. M.; Geller, L. E.; Pechet, M. M. . Journal of the American Chemical Society. 1960, 82 (10): 2640–2641. doi:10.1021/ja01495a061.
- Barton, D. H. R.; Beaton, J. M.; Geller, L. E.; Pechet, M. M. . Journal of the American Chemical Society. 1961, 83 (19): 4076–4083. doi:10.1021/ja01480a030.
- 國際純化學和應用化學聯合會."Barton Reaction".《化学术语总目录》在线版.(英文)
- Nussbaum, A. L.; Yuan, E. P.; Robinson, C. H.; Mitchell, A.; Oliveto, E. P.; Beaton, J. M.; Barton, D. H. R. . The Journal of Organic Chemistry. 1962, 27: 20–23. doi:10.1021/jo01048a004.
- Gutekunst, W. R.; Baran, P. S. . Chemical Society Reviews. 2011, 40 (4): 1976. doi:10.1039/c0cs00182a.
- . Organic Syntheses. 1936, 16: 7. doi:10.15227/orgsyn.016.0007.
- Beckham, L. J.; Fessler, W. A.; Kise, M. A. . Chemical Reviews. 1951, 48 (3): 319–396. doi:10.1021/cr60151a001.
- Sugimoto, A.; Fukuyama, T.; Sumino, Y.; Takagi, M.; Ryu, I. . Tetrahedron. 2009, 65 (8): 1593–1598. doi:10.1016/j.tet.2008.12.063.
- Akhtar, M.; Barton, D. H. R.; Sammes, P. G. . Journal of the American Chemical Society. 1965, 87 (20): 4601–4607. doi:10.1021/ja00948a036.
- Petrovic, G.; Cekovic, Z. . Tetrahedron Lett. 1997, 38: 627–630.
- Barton, D. H. R.; Hesse, R. H.; Pechet, M. M.; Smith, L. C. . Journal of the Chemical Society, Perkin Transactions 1. 1979: 1159. doi:10.1039/P19790001159.
- Akhtar, M.; Pechet, M. M. . Journal of the American Chemical Society. 1964, 86 (2): 265–268. doi:10.1021/ja01056a035.
- Čeković, Ẑ.; Ilijev, D. . Tetrahedron Letters. 1988, 29 (12): 1441–1444. doi:10.1016/S0040-4039(00)80319-6.
- Nickson, A.; Mahajan, J.; McGuire, F. . The Journal of Organic Chemistry. 1961, 26 (9): 3617–3618. doi:10.1021/jo01067a671.
- Čeković, Ž. . Tetrahedron. 2003, 59 (41): 8073–8090. doi:10.1016/S0040-4020(03)01202-X.
- Dorigo, A. E.; McCarrick, M. A.; Loncharich, R. J.; Houk, K. N. . Journal of the American Chemical Society. 1990, 112 (21): 7508–7514. doi:10.1021/ja00177a009.
- Ishmuratov, G. Y.; Kharisov, R. Y.; Shayakhmetova, A. K.; Botsman, L. P.; Shitikova, O. V.; Tolstikov, G. A. . Chemistry of Natural Compounds. 2005, 41 (6): 643–649. doi:10.1007/s10600-006-0003-z.
- Bulliard, M.; Balme, G. V.; Gore, J. . Tetrahedron Letters. 1989, 30 (17): 2213–2216. doi:10.1016/S0040-4039(00)99651-5.
- Robertson, J.; Pillai, J.; Lush, R. K. . Chemical Society Reviews. 2001, 30 (2): 94–103. doi:10.1039/b000705f.
- Akhtar, M.; Barton, D. H. R.; Sammes, P. G. . Journal of the American Chemical Society. 1964, 86 (16): 3394–3395. doi:10.1021/ja01070a039.
- Petrović, G.; Čeković, Ž. . Tetrahedron. 1999, 55 (5): 1377–1390. doi:10.1016/S0040-4020(98)01110-7.
- Allen, J.; Boar, R. B.; McGhie, J. F.; Barton, D. H. R. . Journal of the Chemical Society, Perkin Transactions 1. 1973: 2402. doi:10.1039/P19730002402.
- Barton, D. H. R.; Beaton, J. M. . Journal of the American Chemical Society. 1960, 82 (10): 2641. doi:10.1021/ja01495a062.
- . Journal of the American Chemical Society: 430–431. doi:10.1021/ja00835a039.
- Corey, E. J.; Hahl, R. W. . Tetrahedron Letters. 1989, 30 (23): 3023–3026. doi:10.1016/S0040-4039(00)99392-4.
- Dehaen, W.; Mashentseva, A. A.; Seitembetov, T. S. . Molecules. 2011, 16 (12): 2443–2466. doi:10.3390/molecules16032443.
- László Kürti, Barbara Czakó: Strategic Applications of Named Reactions in Organic Synthesis; Elsevier Academic Press, Burlington-San Diego-London 2005, 1. Edition; ISBN 0-12-369483-3.
