化学动力学
化学动力学也称反应动力学、化學反應動力學,是物理化学的一个分支,研究化学反应的反应速率及反应机理。它的主要研究领域包括:分子反应动力学、催化动力学、基元反应动力学、宏观动力学、表观动力学等,也可依不同化学分支分类为有机反应动力学及无机反应动力学。化学动力学往往是化工生产过程中的决定性因素。
化学动力学与化学热力学不同,不是计算达到反应平衡时反应进行的程度或转化率,而是从一种动态的角度观察化学反应,研究反应系统转变所需要的时间,以及这之中涉及的微观过程。化学动力学与热力学的基础是统计力学、量子力学和分子运动论。
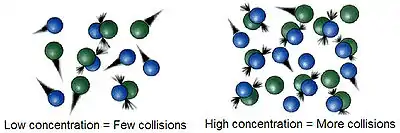
反应速率
反应速率是化学反应快慢程度的量度,广义地讲是参与反应的物质的量随时间的变化量的绝对值,分为平均速率与瞬时速率两种。平均速率是反应进程中某时间间隔(Δt)内参与反应的物质的量的变化量,可用单位时间内反应物的减少量或生成物的增加量来表示;瞬时速率是浓度随时间的变化率,即浓度-时间图像上函数在某一特定时间的切线斜率。
用实验方法测定反应速率受反应物量或浓度影响的定量方程称为速率方程,速率方程中各浓度的指数称为相应物质的反应级数,它们的和称为“总反应级数”,也可简称“反应级数”。反应的级数是不能由化学方程式的类型推断的,常见的级数反应如零级反应、一级反应、二级反应,都会有各自不同的速率方程。速率方程中的常数k称为速率常数,可看作单位浓度下的反应速率。建立速率方程时一般采取两种方法:作图法与初速法。
控制化学反应速率是许多实践活动的需要。绝大多数化学反应的速率都是随着反应进行而不断减慢的,并且同一反应,在不同浓度、温度、压力、相态下,是否使用催化剂以及使用不同的催化剂,都会对反应速率造成不同的影响。
影响速率的因素
反应物性质
化学反应的速率自然与反应物的结构与性质有着很大的关系。爆炸反应、强酸与强碱的中和反应以及离子交换反应的速率非常快,但岩石的风化、钟乳石的生长以及铀的衰变等,却需要千百万年才有显著的变化。一般而言,生成共价键和大分子的反应速率比较慢。
有些热力学可行的反应,在动力学上却因为速率太慢而几乎不发生,如常温下氢氧化合生成水,金刚石在常温常压下转化为石墨等。一般限制这些反应发生的因素,称为动力学因素,可归咎于反应物的结构、化学键的类型及过渡态方面。
状态
反应物的状态(固态、液态、气态、溶液态)对反应速率也有很大的影响。一般而言,速率决定于反应物接触表面积与体积的比值,该比值越大,反应速率越快。因此,均相反应中反应物接触较充分,速率较快,如水溶液中的中和反应及沉淀反应;异相反应中的反应物只限于在接触面反应,速率较慢,且一般需要剧烈摇晃或搅拌容器,以充分混合使反应物接触面积增大。固体为粉末状或块状对反应也有影响,粉末状的固体接触表面积较大,反应较快。
有机化学中的水相反应是异相反应比均相反应快的一个例子。
浓度
反应物及生成物的浓度也会在很大程度上左右化学反应速率。根据碰撞理论,反应物浓度增加时,反应中的分子碰撞频率——即“频率因子”增加,反应速率加快。而根据速率方程,反应速率r与一个或多个反应物或其指数的浓度成正比,因此增加反应物浓度无疑会使反应速率增大。 对于反应级数为负数的反应,增加反应物浓度则会使反应速度降低。

温度
温度升高,化学反应的速率增大,无论是放热反应还是吸热反应,都是如此。分子在高温时热能及振动能增大,与其他反应物碰撞的频率也会随此增多,使化学反应加快。粗略计算温度对反应速率的影响时,常会提到阿伦尼乌斯方程,形式如下:

该式中的是速率常数,是温度,是气体常数,和是两个参数,分别称为“指前因子”和“活化能”。指前因子和活化能的值随温度而变化,但阿伦尼乌斯对它们采取线性化处理,以简化该方程。事实证明,大多数反应在一定温度区间作该近似是完全允许的。





温度对速率常数的影响在于后的指数项,化学反应速率随温度升高而增大的快慢,与它的值有关。同一温度区间内,越小的反应,温度升高速率增大得越快,值增大会使温度升高速率减慢。由于该方程是指数函数,因此T的微小变化会使速率常数发生很大的变化。
催化剂
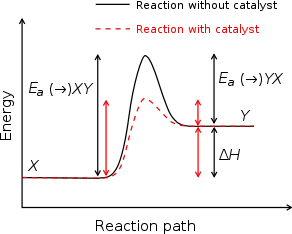
催化剂是可以改变反应速率但在反应前后并不改变组成的物质,通常不包括反应溶剂。根据催化剂的状态,可将其分类为均相催化剂和异相催化剂;从催化剂加快还是减慢反应速率的角度,可把催化剂分类为正催化剂和负催化剂两大类。过渡态理论认为,催化剂之所以可以加快反应速率,是因为它改变了反应途径,生成了不同的活化络合物而降低了活化能。催化剂不会影响达到平衡时的各组分浓度,它影响的只是达到平衡的速度快慢。
搅拌溶液会增加微粒的动能,增加反应物之间的碰撞频率,从而使反应速率增大。
有气体参与的反应中,增加气体的分压也会使反应物之间的碰撞频率增大,气体的活度增加,从而使化学反应速率变快。
此外,过渡态理论认为活化熵也是影响化学反应速率的因素。若将化学反应过程比作翻越山岭,那么活化能类似于山峰的高度,活化熵则是找到爬山方位的容易程度。其值越大,代表越容易找到爬坡的方位,反应物越容易被转化为活化络合物。有些蛋白酶催化的蛋白水解反应中,虽然用酶作催化剂时的活化能要比用酸催化时高出12kJ/mol,但酶催化的速率仍然要高得多。这就可以用活化熵的概念来解释。
實驗方法
量測反應速率的實驗方法會量測反應物及生成物濃度隨時間的變化。例如,反應物的濃度可用光譜光度測定法量測,針對只受反應物影響,系統其他反應物或生成物都不會吸收其能量的波長來進行行量測。
若是針對需要幾分鐘才會進行的反應,可以讓反應物都先到達需要的溫度,再混合反應物啟動反應,並且開始量測。

反应平衡
热力学研究反应达到反应平衡时的状态。在可逆反应中,反应物与产物达到动态平衡,正向反应与逆向反应的速率相等,反应物与产物的浓度不再发生变化。它可通过哈伯法合成氨、化学振荡反应如Belousov-Zhabotinsky反应(B-Z反应)、碘钟反应等多组分反应过程来进行演示。
反应机理
虽然化学方程式中各物质的计量比看似简单,但微观上,一个化学反应通常是经过几步完成的,描述化学反应的微观过程的化学动力学分支称为反应机理。反应机理中,每一步反应称作基元反应,基元反应中反应物的分子数总和称为反应分子数。反应机理由一个或多个基元反应所组成,这些基元反应的净反应即为表观上的化学反应。
这些基元反应中,反应速率最慢的一个称为速率控制步骤,简称“速控步”,是决定总反应的速率的步骤。正如从酒瓶中倒酒时,决定酒的流速的是瓶颈的直径一样,速率控制步骤对于整个化学反应的速率也有类似的效应,称为“瓶颈效应”。通常情况下,速控步后的任何基元反应对反应速率是丝毫没有贡献的。
为了由提出的反应机理推导出实验得到的速率方程,一般采取两种方法:平衡假设法和稳态近似法。前者方法中,假设互逆反应在反应中可以达到平衡,然后推出反应的速控步,进一步得到速控步的速率方程,并将该方程中不属于计量方程式的反应物用其他表达式代替;后者方法中,假设中间产物的浓度在反应过程中保持近似不变,并假设某一步为速控步,推导出最后的表象速率方程。
参考资料
- Keith J. Laidler, K. J. Chemical Kinetics (3rd ed., Harper and Row 1987) p.33-39 ISBN 0-06-043862-2
- Espenson, J.H. Chemical Kinetics and Reaction Mechanisms (2nd ed., McGraw-Hill 2002), p.254-256 ISBN 0-07-288362-6
- Atkins P. and de Paula J., Physical Chemistry (8th ed., W.H. Freeman 2006) p.793 ISBN 0-7167-8759-8
- Laidler, K.J. Chemical Kinetics (3rd ed., Harper and Row 1987) p.359-360 ISBN 0-06-043862-2
- Espenson, J.H. Chemical Kinetics and Reaction Mechanisms (2nd ed., McGraw-Hill 2002), p.264-6 ISBN 0-07-288362-6
- Steinfeld J.I., Francisco J.S. and Hase W.L. Chemical Kinetics and Dynamics (2nd ed., Prentice-Hall 1999) p.94-97 ISBN 0-13-737123-3
- Espenson, J.H. Chemical Kinetics and Reaction Mechanisms (2nd ed., McGraw-Hill 2002), p.256-8 ISBN 0-07-288362-6
- Steinfeld J.I., Francisco J.S. and Hase W.L. Chemical Kinetics and Dynamics (2nd ed., Prentice-Hall 1999) p.140-3 ISBN 0-13-737123-3
- Atkins P. and de Paula J., Physical Chemistry (8th ed., W.H. Freeman 2006) pp.805-7 ISBN 0-7167-8759-8
- Preparing for the Chemistry AP Exam. Upper Saddle River, New Jersey: Pearson Education, 2004. 131-134. ISBN 0-536-73157-8
- 北京师范大学,华中师范大学,南京师范大学无机化学教研室编。《无机化学》第四版上册。北京:高等教育出版社,2002年。ISBN 7-04-010768-6