诱导性多能干细胞
诱导性多能干细胞(英語:),又稱人工誘導多能幹細胞,常簡稱爲iPS細胞(iPSC),是一種由哺乳動物成體細胞經轉入轉錄因子等手段脫分化形成的多能幹細胞,最早由日本學者山中伸彌的研究團隊於2006年發現[1]。山中伸彌團隊在發表iPS誘導技術時使用實驗材料爲小鼠細胞。2007年,研究人員又證明iPS誘導技術可以應用於人體細胞[2]。最初由山中伸彌團隊發現的誘導方法是通過慢病毒載體將Oct4、Sox2、c-Myc、Klf4四種轉錄因子基因轉入成體細胞將其轉化爲類似於胚胎幹細胞的多能幹細胞。其後,研究人員又先後發現了更優化的誘導方法,如使用質粒載體轉染、腺病毒感染、脂質體導入等非基因組整合的方法進行誘導[3]、通過細胞融合誘導[4]、使用小分子藥物進行誘導[5]、轉入miRNA(微干擾RNA)進行誘導[6]等。

iPS細胞與胚胎幹細胞擁有相似的再生能力,理論上可以分化爲成體的所有器官、組織。而相比胚胎幹細胞,iPS細胞面臨的倫理道德爭議較小,且應用該技術可以產生基因型與移植受體完全相同的幹細胞,規避了排異反應的風險,因而iPS細胞在一定程度上衝擊了胚胎幹細胞在再生醫學中的地位,被認爲在再生醫學及組織工程方面擁有較爲廣闊的應用前景,有望爲治癒糖尿病、關節炎等疾病提供新的思路。同時,iPS細胞在新藥開發、疾病模型構建領域也有望得到應用。但iPS誘導技術同樣面臨着誘導效率低、用於治療可能存在長期風險等挑戰[7][8][9][10]。
製備方法
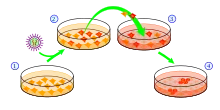
最初由山中伸彌團隊發現的iPS細胞製備(誘導)方法是以通過慢病毒載體轉入數個轉錄因子爲核心,在導入四種轉錄因子後,小鼠的成纖維細胞經過一定時間就會轉變爲狀態類似於胚胎幹細胞的iPS細胞[1]。
使用這種方法製備iPS細胞,首先需要一個特殊的轉基因小鼠品系。這種轉基因小鼠的Fbx15基因下游轉入了一個βgeo元件。該元件由β-半乳糖苷酶基因和新黴素抗性基因(NeoR)融合而成。如果基因表達環境與胚胎幹細胞相似,Fbx15基因就會表達。而在一般的成體細胞中,Fbx15基因表達處於關閉狀態。對這種品系的轉基因小鼠來說,細胞如果處於一種與胚胎幹細胞相似的狀態,Fbx15基因下游的βgeo元件也隨Fbx15基因會一同表達,βgeo元件中的NeoR基因會使這種細胞獲得對藥物G-418的抗性,而一般的體細胞仍然對G-418敏感。加入G-418後,對G-418敏感的成體細胞會死亡,而與胚胎幹細胞狀態相似的細胞因具有G-418抗性,會在篩選中存活[1][12]。
後續實驗用到的小鼠胚胎成纖維細胞(MEF)或尾尖成纖維細胞(TTF)均來自上述的轉基因小鼠品系。在得到該品系小鼠的成纖維細胞後,需要製備分別含有Oct4、Sox2、c-Myc、Klf4基因的四種慢病毒載體。之後,再用這四種慢病毒載體感染成纖維細胞。用於實驗的成纖維細胞已與有G-418抗性的胚胎幹細胞飼養層細胞混合,以維持可能會在後續實驗中產生的iPS細胞的幹性。如果感染成功,這四種基因就會在成體細胞中表達。感染後,需要將細胞培養基更換爲含有G-418的胚胎幹細胞培養基。細胞如果回到類似胚胎幹細胞的狀態,會因NeoR基因的表達於含G-418的培養基中存活,而未回到類似胚胎幹細胞狀態的成纖維細胞會死亡。飼養層細胞因帶有G-418抗性,也會在這個過程中存活。經過7天左右,可以觀察到有細胞形成類似胚胎幹細胞的細胞集落,這些細胞就是誘導產生的iPS細胞。再經過一段時間的生長,待細胞集落足夠大時,即可挑選合適的集落進行轉移,經過多代培養後形成穩定的iPS細胞系[1][13]。
在山中伸彌於2006年發表關於iPS誘導技術的文章後,2007年,研究人員成功將iPS技術應用於人成體細胞,製得人源性的iPS細胞,方法與山中伸彌團隊的製備方法有少許不同[8][14]。其後,研究人員又先後成功製備了山羊、綿羊、大鼠、豬、貓、兔、狗、狼等哺乳動物的iPS細胞[4]。同時,亦發現除成纖維細胞外,其他類型的成體細胞以及成體幹細胞均可以重編程爲iPS細胞[8][15][16][15]。不過,不同細胞重編程到iPS細胞的效率存在差異。一般來說,分化程度越低的細胞越容易被重編程爲iPS細胞[17][18][19]。同時,研究人員也對重編程的方法進行了一定改良。比如,已有使用腺相關病毒載體感染、質粒載體轉染、脂質體轉入等非基因組整合技術爲核心的重編程方法[3]。亦有研究表明,只通過轉入特定miRNA(微干擾RNA)就可以使細胞重編程爲iPS細胞[6]。將成體細胞與胚胎幹細胞的細胞質融合,也可以使其重編程爲iPS細胞[4]。只通過加入多種小分子藥物的混合物,亦可達到將成體細胞重編程爲iPS細胞的目的[5]。一些實驗結果表明,通過改變轉入的轉錄因子等方法,可以在一定程度上提高iPS的重編程效率[4]。
性質
three_germ_line_cells_differentiated_from_iPSCs.png.webp)
iPS細胞性質與胚胎幹細胞相似,但在一些方面又存在差異[8][1][20]。培養iPS細胞的環境與胚胎幹細胞相似。傳統的培養方法是將iPS細胞培養在經絲裂黴素或射線滅活的小鼠胚層成纖維細胞(MEF)組成的飼養層(feeder)上,並使用含有血清及白血病抑制因子(LIF)的培養基中[1][13]。目前亦已有方法可以將iPS細胞培養在化學成分明確的無血清培養基上,且不需要使用飼養層細胞的培養方法[21]。
iPS細胞在體外具有無限增殖的潛能,也能形成與胚胎幹細胞相似的緊緻、平坦的細胞集落。iPS細胞在體外培養時,形態也與胚胎幹細胞接近:細胞呈圓形,細胞核體積大、細胞質體積相對較小。同時,iPS細胞也表達一些胚胎幹細胞中的幹細胞標誌物,比如Nanog蛋白、SSEA類蛋白、TRA類蛋白[8][1]。iPS細胞具有分化爲三個胚層細胞或組織的潛力[1][22]:76-77。iPS細胞在注射入免疫缺陷性個體後,可以生成畸胎瘤[22]:321,體外懸浮培養的iPS會分化形成類胚體(embryoid body,EB)[23]。但iPS形成嵌合體(chimera)的能力較差,甚至使用iPS細胞產生嵌合體小鼠的嘗試曾一度失敗[22]:321[20]。此外,iPS細胞形成的細胞集落是異質性的,一部分iPS細胞集落中的細胞與胚胎幹細胞存在較大差異,可從形態和是否表達Dlk1-Dio3等標誌物將這個集落區分出來。從形態正常的細胞集落中挑選的iPS細胞的基因表達模式和胚胎幹細胞基本相似,但亦存在一定差異。分析表明,一些基因的表達情況在iPS細胞中和胚胎幹細胞中存在持久性的差異[20]。
歷史

1950年代,英國發育生物學家約翰·格登的一系列實驗表明,將蟾蜍成體細胞的細胞核移入去除細胞核的卵細胞後,這個重組的細胞可以發育爲一個完整的蟾蜍個體。這一發現否定了此前一度流行的一個學說:細胞在分化的過程中會不斷丟棄不需要的遺傳物質。約翰·格登的實驗證明動物成體細胞仍然擁有全套基因組,有發育成完整個體的潛力[24]:5-6[9]。
iPS細胞誘導技術的發現者山中伸彌於1999年入職日本奈良先端科學技術大學院大學(NAIST)擔任副教授。在1999年至2003年間,他提出了通過轉入外源性因子使體細胞重編程爲幹細胞的猜想,並與博士後高橋和利開始了相關的研究[25]。2003年,山中伸彌得到了大阪大學教授岸本忠三的支持,獲得科学技術振興機構5年3億日圓的經費支持。同年,山中伸彌升爲正教授[25]。2004年,山中伸彌轉任京都大學教授[26]。
山中伸彌團隊最初選擇了24個候選基因,並製備了分別含有這24個候選基因的慢病毒載體。最初,山中伸彌的團隊同時將這24種慢病毒載體轉入小鼠的成纖維細胞中,發現有部分細胞回到了與胚胎幹細胞類似的狀態,並形成了細胞集落。在經過幾次重複實驗確認結果的可靠性後,山中伸彌團隊又在這24種基因中進行了進一步篩選,最終確認了一組最佳的組合:當同時向成體細胞轉入Oct4、Sox2、c-Myc、Klf4四種轉錄因子時,就能有細胞轉化爲多能性幹細胞。2006年,山中伸彌團隊發表了他們的這一實驗結果[27][24]:15-17。在山中伸彌發表這篇論文後,實驗結果的真實性曾一度遭到同行的質疑。2007年4月,美國生物學家鲁道夫·耶尼施首次表示山中伸彌的實驗是可重複的。隨後,其他課題組也先後重複出山中伸彌的實驗[24]:17-19。同年,研究人員成功使用人的成體細胞取得人源性的iPS細胞[8]。之後,研究人員又成功製備了山羊、大鼠、狗等哺乳動物的iPS細胞[4]。
在山中伸彌團隊宣佈發現iPS細胞後,iPS細胞很快成爲生命科學領域研究的熱門。根據統計,僅2006年到2009年之間,就有300餘篇關於iPS細胞的論文發表[27]。iPS細胞誘導技術的發現者山中伸彌於2012年與約翰·格登爵士一同獲得了諾貝爾生理醫學獎[11]。同年,京都大學教授高橋政代與山中伸彌合作,計劃展開一項使用iPS細胞治療黃斑部退化的臨床試驗。在該試驗中,會先用患者的體細胞產生iPS細胞,然後再令取得的iPS細胞分化爲视网膜色素上皮細胞。最後,將得到的视网膜色素上皮細胞用於修復患者的視網膜。2014年,該計劃正式進入臨床試驗階段,成爲全球首個進入臨床試驗階段的iPS細胞相關治療方案。試驗開始後,研究團隊曾一度宣佈實驗進展順利,患者的病情得到了緩解,視力也有了提高。但最後因爲發現iPS細胞和分化的細胞基因組中存在兩處變異,研究團隊於2015年宣佈停止這項臨床試驗[27][28]。2016年3月,高橋政代團隊又進行了一次移植手術,將重編程自正常人體細胞的iPS細胞重新分化爲视网膜色素上皮細胞,並將之移植入一名黃斑部退化患者的眼部。移植手術本身是成功的,但後續報導表明,患者在手術後產生了嚴重的不良反應[29][30][31]。
挑戰與應用前景
Ips_cells.png.webp)
iPS細胞性質與胚胎幹細胞相似,且相比胚胎幹細胞會面臨較少的倫理學爭議,因而iPS細胞被認爲在組織工程及再生醫學、藥物開發、疾病模型構建等領域有較廣闊的發展前景[8][9]。但另一方面,iPS細胞的誘導技術仍有一些不成熟之處,iPS細胞在得到真正的臨床應用前,還需要解決一些關鍵性的問題[8][20][32]。
挑戰
iPS誘導技術處於快速發展時期,還有許多不夠成熟的地方[8]。首先,iPS細胞的部分性質與胚胎幹細胞存在差異,如iPS形成嵌合體的能力較弱[22]:321[20]。此外,iPS相比胚胎幹細胞較難分化爲成體細胞[19]。因爲誘導iPS細胞需要人爲導入外源性因子,細胞的表觀遺傳狀態也可能會因此出現異常。如果應用iPS細胞作爲細胞替代療法的材料,可能會存在長期的風險[22]:6-7:78。有研究表明,iPS細胞的成瘤性比胚胎幹細胞強許多[33]。另一方面,iPS細胞的誘導技術還面臨誘導效率低的問題。一般認爲這是因爲體細胞重編程到多能幹細胞的過程中,會遇到一些「屏障」。比如,導入的基因難以與表達處於關閉狀態的下游的基因發生相互作用[8][19]。簡而言之,如果想要將iPS細胞用於臨床療法中,需要滿足以下幾個條件:確保能高效、安全地取得iPS細胞;確保iPS細胞能分化爲目的細胞;確保病人不會產生不良反應[19][32]。另外,來自不同細胞集落的iPS細胞間存在差異。如果想要將iPS細胞應用於新藥開發中,就必須要想辦法控制這些差異,以保證不同批次的實驗組間不存在顯著的無關變量[19]。
應用前景
iPS細胞因爲可以用於細胞替代療法而受到關注[24]:20-22。目前,已成功將iPS分化成了來自三個胚層的不同細胞和組織[10]。另外,已成功在小鼠體內用iPS細胞修復了受損的視網膜和血管[34][35]。研究人員對基於iPS細胞的細胞替代療法的設想是,使用病人的成體細胞產生與病人基因型一致的iPS細胞,再於體外誘導產生所需的器官,最後通過移植手術將產生的器官植入病人體內,使病人機體受損的功能得以恢復。因爲使用的細胞基因型與病人相同,移植手術後理論上不會產生排異反應[8][19][32]。利用這樣的細胞替代療法,有望治癒因細胞受損而產生的疾病,比如由胰島B細胞受損引發的1型糖尿病、由血管內皮細胞受損引發的心血管疾病等。同時,亦可用來產生移植手術所需要的器官[19][32][10]。美國國防部已批准了包括用iPS細胞產生血液的研究在內的幾項基金。有設想認爲可以用iPS構建不同血型的iPS細胞庫,爲戰爭傷員或病人源源不斷供應血液[24]:20-22。
iPS細胞亦可以應用於新藥開發中。比如,可以用iPS細胞在體外分化出成體細胞,再用藥物處理分化的成體細胞,用以預測這些藥物在實際應用到體內後是否會令使用者產生不良反應[27][32][19][36]。此外,亦可以使用iPS細胞進行疾病模型的構建。即用病人的iPS細胞構建狀態異常的成體細胞或組織。這樣的成體細胞或組織可用作相關疾病的研究模型。目前,研究人員有計劃用iPS細胞建立一個疾病模型庫[37][38][39]。
参考文献
- Takahashi, K; Yamanaka, S. . Cell. 2006, 126 (4): 663–76. PMID 16904174. doi:10.1016/j.cell.2006.07.024.

- Takahashi, K; 等. . Cell, 131(5), 861–872. 2007.
- Yi-ye Zhou; Fanyi Zeng. . Genomics Proteomics Bioinformatics. 2013 Oct; 11(5): 284–287.
- Jong Soo Kim; 等. . Int J Stem Cells. 2011 Jun; 4(1): 1–8.
- Xiaojie Ma; Linghao Kong; Saiyong Zhu. . Protein Cell. 2017 May; 8(5): 328–348.
- Anokyedanso, Frederick; 等. . Cell Stem Cell 8.4(2011):376.
- Harvey Lodish; 等. . . Macmillan Higher Education. 2013: 979–983. ISBN 978-1-4641-0981-2.
- Robert Lanza; 等. . San Diego: Elsevier. 2009: xxi–xxii. ISBN 978-0-12-374729-7.
- Kazutoshi Takahashi; Shinya Yamanaka. . Development 2013 140: 2457-2461.
- Vimal Selvaraj; 等. . Trends Biotechnol. 2010 Apr; 28(4): 214–223.
- . NobelPrize.org. 2012-10-08 [2012-10-08]. (原始内容存档于2017-04-26).
- Yanhong Shi. . Curr Mol Pharmacol. 2009 Jan; 2(1): 15–18.
- Kazutoshi Takahashi; 等. . Nature Protocols. 2007, 2 (12).
- In-Hyun Park; 等. . Nature Protocols. 2008, 3 (7).
- Melissa Helen Little. . Elsevier Science. 2015-08-06: 493–. ISBN 978-0-12-800438-8. (原始内容存档于2018-02-07).
- Jeong Beom Kim; 等. . Nature Protocols volume4, pages1464–1470 (2009).
- Niibe Kunimichi; 等. . Plos One 6.3(2011):e17610.
- J Yee. . Nature Education. 2010, 3 (9): 25. (原始内容存档于2017-05-30).
- Charles A. Goldthwaite. . NIH. (原始内容存档于2017-10-29).
- Bilic J; Izpisua Belmonte JC. . Stem Cells. 2012 Jan;30(1):33-41.
- Guokai Chen; 等. . Nat Methods. 2011 May; 8(5): 424–429.
- Tarik Regad; 等. . Wiley Blackwell. 2015: 3–5. ISBN 978-1-118-67062-0.
- Yongshun Lin; Guokai Chen. . Stembook.
- 內莎·凱里(Nessa Carey)著 賈乙、王亞菲譯. . 重慶出版社. 2011. ISBN 978-7-229-10427-6.
- . 毎日新聞. 2012-10-08 [2012-10-08]. (原始内容存档于2012-10-10).
- . nobelprize.org. 2014. (原始内容存档于2018-01-27).
- Megan Scudellari. . Nature News&Reviews. 2016. (原始内容存档于2018-01-09).
- Garber, Ken. . Nature Biotechnology: 890–891. PMID 26348942. doi:10.1038/nbt0915-890.
- Takahashi M. . Nippon Ganka Gakkai Zasshi. 2016 Mar;120(3):210-24; discussion 225.
- . RIKEN Center for Developmental Biology. 2017-04-04 [2017-09-06]. (原始内容存档于2017-04-18).
- . The Japan Times. 2018-01-17. (原始内容存档于2018-01-27).
- . . NIH. (原始内容存档于2018-01-22).
- Ivan Gutierrez-Aranda.; 等. . Stem Cells. 2010, 28 (9): 1568–1570. PMC 2996086. PMID 20641038. doi:10.1002/stem.471.
- Mullin, Emily. . fiercebiotech.com. 2014-01-28 [2014-02-17]. (原始内容存档于2014-02-22).
- Zambidis, Elias; Lutty, Gerard; Park, Tea Soon; Bhutto, Imran; 等. . Circulation (American Heart Association). 2014, 129 (3): 359–372. PMC 4090244. PMID 24163065. doi:10.1161/CIRCULATIONAHA.113.003000. (原始内容存档于2014-02-27).
- Shinnawi, Rami; Huber, I; Maizels, L; Shaheen, N; Gepstein, A; Arbel, G; Tijsen, A; Gepstein, L. . Stem Cell Reports. 2015, 5 (4): 582–596. PMC 4624957. PMID 26372632. doi:10.1016/j.stemcr.2015.08.009.
- Grskovic, M; Javaherian, A; Strulovici, B; Daley, GQ. . Nature Reviews. Drug Discovery. 2011-11-11, 10 (12): 915–29. PMID 22076509. doi:10.1038/nrd3577.
- Grskovic, M; Javaherian, A; Strulovici, B; Daley, GQ. . Nature Reviews. Drug Discovery. 2011-11-11, 10 (12): 915–29. PMID 22076509. doi:10.1038/nrd3577.
- Gerlin, Andrea. . Bloomberg.com. 2012-12-05. (原始内容存档于2017-02-11).
外部連結
- 京都大學iPS研究所 页面存档备份,存于
