長期增強作用
長期增強作用(英語:,LTP)又稱长时程增强作用、長期增益效應,是由于同步刺激两个神经元而发生在两个神经元信号传输中的一种持久的增强现象。[2]这是与突触可塑性——突触改变强度的能力相关的几种现象之一。[3]由于记忆被认为是由突触强度的改变来编码的,LTP被普遍视为构成学习与记忆基础的主要分子机制之一。[2][3]
LTP是1966年泰耶·勒莫在兔海马体中发现的,一直以来是研究的热门主题。许多现代的LTP研究试图更好地了解其生物学基本原理,而其他一些研究则以探索LTP和行为学习之间的因果关系为目标。还有一些则试图开发通过提高LTP改善学习和记忆的方法,不管是采用药物手段还是其他手段。LTP还是临床研究的主题,比如在阿兹海默病和成瘾医学领域。
研究史
早期学习理论

在19世纪末,科学家们普遍认为成人大脑的神经元的数量(约1000亿个[4])不会随年龄增长而显著增加,因此神经生物学家一般都相信记忆的形成并非神经元增生的結果[5]。由于这种认识,记忆如何在没有新神经元形成时产生,就成为一个有待解决的问题。
西班牙神经解剖学家圣地亚哥·拉蒙-卡哈尔是最早认识到学习机制并不需要形成新神经元的科学家之一。在他于1894年在伦敦皇家内科医学院发表的演讲中,他提出,记忆可能是由加强现有神经元之间的联系、从而提高它们沟通的有效性而形成的。[5]唐纳德·赫布在1949年提出的理论中呼应了卡哈尔的思想,进一步提出细胞可能通过构建新的连接或经历代谢变化而提高它们的沟通能力。
让我们假设,反射活动的持续或重复(“跟踪”)往往诱发持久的细胞变化,加诸其稳定性之上……当细胞A的一个轴突距细胞B足够近,能够反复或持续的向其发射电信号,一些生长过程或代谢变化就会使得至少一个细胞——比如细胞A的效率,得到增强。[6]
虽然这些记忆形成的理论现在已是常识,但当时却是超前的。19世纪末和20世纪初的神经科学家和心理学家還没有足夠的神经电生理学技术,能觀察神經元內電位的微電極一直到1949年才發明[7],长时程增强作用就是在这之後才发现的[8]。
发现
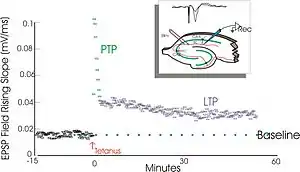
泰耶·勒莫1966年首次在挪威奥斯陆佩尔·安德森的实验室中观察LTP。[8][9]在那儿勒莫对经过麻醉的兔进行了一系列神经生理学实验,以研究海马体在短期记忆中的作用。
勒莫的实验聚焦于神经节点、或突触,从穿通纤维到齿状回。勒莫通过刺激穿通通路的突触前纤维和记录齿状回突触后细胞的反应来进行这些实验。正如预期的那样,单脉冲电信号刺激穿通通路纤维引发了齿状回细胞的兴奋性突触后电位(excitatory postsynaptic potential,EPSP)。勒莫意外的观察到,当他对突触前纤维施加高频度刺激时,突触后细胞对这些单脉冲刺激的反应会增强很长一段时间。当这一系列刺激被接受后,后续的单脉冲刺激会在突触后细胞群中激发增强、延长了的EPSP。这种现象——即高频刺激可引发突触后细胞的持久增强反应——最初被称为“持久增强作用”(long-lasting potentiation)。[10][11]
蒂莫西·布利斯1968年加入了安德森的实验室,[8]与勒莫合作,二人在1973年发表了第一篇关于海马体长时程增强作用的论文。[10]布利斯和托尼·加德纳-梅德温在同一期刊物中发表了在清醒动物身上观察到长时程增强效应的类似报告。[11]1975年,道格拉斯和戈达德提出将“长时程增强”作为持久增强作用的新名称。[12]安德森建议发现者采纳这个新名词,也许是因为其缩写“LTP”更容易发音。[13]

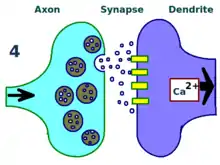
种类
虽然LTP最初是在兔海马体中发现的,但后来在其他神经结构——比如大脑皮质、小脑、杏仁核等组织中都发现了这种现象。[15]罗伯特·马伦卡,一个著名的LTP研究者,认为LTP甚至可能发生在所有哺乳动物大脑中的兴奋性突触。[16]
不同的大脑区域表现出不同形式的LTP。神经元之间的特殊LTP类型取决于多项因素。其中一个因素观察LTP时被观察者的年龄。例如,未成熟海马体中LTP的分子机制不同于那些成熟海马体LTP的机制。[17]一个特定的细胞所使用的信号转导通路与LTP的特定类型有关。例如,一些海马体的LTP类型取决于NMDA受体,其他一些类型则可能取决于代謝型麩胺酸鹽受體(mGluR),还有一些则取决于其他分子。大脑中信号通路的这些于LTP关联的不同类型,以及各种通路在脑中的广泛分布,决定了不同的神经元间的LTP部分取决于观察到它的解剖部位。[16]例如,海马体谢弗侧支通路中的LTP是NMDA受体依赖型,而苔藓纤维通路中的LTP则不依赖NMDA受体。[18]
诱导LTP所需的突触前和突触后活动是LTP分类的又一标准。广义上讲,LTP可以分为成赫布机制、非赫布机制和反赫布机制。这些名词借自赫布的假设,总而言之就是“互相发射电信号的细胞连接在一起”。诱导赫布型LTP需要突触前和突触后同步去极化。[19]非赫布型LTP则不需要突触前和突触后同时去极化,海马体苔藓纤维通路中的LTP就是一个例子。[20]反赫布型LTP是非赫布型LTP的一种特殊类别,诱导它要求同步的突触前去极化和相应的突触后超极化。[21]
由于其组织可见、LTP诱导容易操作,海马体CA1区已成为研究哺乳动物LTP的典型位点。特别是成熟海马体CA1区NMDA受体依赖型LTP是LTP中研究的最多的类型,也是本条目的重点。[16]
性质
NMDA受体依赖型LTP具有几个特性,包括输入专一性、关联性、协同性和持久性。
- 输入专一性
- 一个突触的LTP一经诱导,不会扩散到其他突触,因而LTP具有输入专一性。LTP传播到那些依据关联性和协同性法则所规定的突触。但是,LTP的输入专一性法则在短距离内不一定特别精确。弗雷和莫里斯在1997年提出了一种解释输入专一性的假说,即突触标识和捕获假说。[22]
- 关联性
- 关联性是指,当一条通路的弱刺激尚不足以诱导LTP时,另一通路的强刺激会同时诱导两条通路的LTP。[22]
- 协同性
- LTP可由强烈的强直刺激激发突触的单一通路,或通过许多较弱的刺激协作引发。当一条通向突触的路径受到弱刺激,它产生的突触后去极化不足以诱导LTP。与此相反,当微弱的刺激施加到许多通路,而这些通路均汇聚到一片单一的突触后膜时,产生个别性突触后去极化可以共同突触后细胞去极化,足以诱导LTP的合作。突触标识可能是关联性与协同性的共同基础。布鲁斯·麦克诺顿认为,关联性和协同性之间的差别仅仅是语义上的。[23]
- 持久性
- LTP的作用时间是持久的,可以持续几分钟乃至几个月。这是它与其他突触可塑性的根本区别。[24]

维持
由于诱导LTP需要短期的激发钙调蛋白激酶Ⅱ和蛋白激酶C,维持E-LTP(LTP的早期形式)的一个特点就是这些酶的持续活动。在这一阶段,与钙不相联系的ζ型蛋白激酶C,会自发的活跃起来。最终它们就能引发磷酸化事件,这是E-LTP所必须的。[26]
表达
在磷酸化反应中,一个小的磷酸基团被添加到另一个分子,以改变那个分子的活性。自动激发的CaMKII和PKC使用磷酸化反应来进行E-LTP表达的两大主要机制。首先,最重要的是,现有的AMPA受体磷酸化可以增强活性。其次,它们介导或调节额外的AMPA受体插入到突触后膜。[16]重要的是,在E-LTP中分配AMPA受体给突触与蛋白质合成过程是各自独立的。这是因为突触后膜上连接有非突触的AMPA受体储存。适当LTP诱导的刺激到达时,非突触AMPA受体迅速在蛋白激酶的作用下募集到突触后膜上。如前所述,AMPA受体是大脑的最丰富的谷氨酸受体,可调节其大部分兴奋活动。通过增加突触中AMPA受体的效率和数量,后续的兴奋性刺激就能产生更大的突触后反应。[27]
上述E-LTP的模型描述的全部是诱导、维持和表达的突触后机制,不过E-LTP的表达可能会出现一个附加的突触前程序。其中一种假说认为,这种突触前促进程序是由于E-LTP过程中突触后细胞中持久的CaMKII活动可能会导致一种“逆行信使”的合成。根据这一假说,新合成的信使穿越突触后细胞与突触前细胞间的突触间隙,而导致促进对后续刺激的突触前反应的一系列事件。这些事件可能包括神经递质囊泡数量或囊泡释放概率的增加,或两者兼而有之。逆行信使除了为突触前早期LTP表达打下基础,也可能在后期LTP中发挥作用。[28]
后期
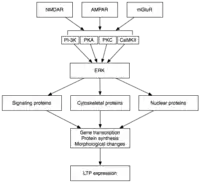
后期LTP(L-LTP)是E-LTP的自然延伸。不同于与蛋白质合成相互独立的E-LTP,L-LTP需要基因转录[29]和蛋白质合成,[30]这些过程均发生在突触后细胞中。L-LTP有两个阶段:第一阶段依赖于蛋白质合成,第二阶段则既需要基因转录,也需要蛋白质合成。这两个阶段有时分别被称为LTP2和LTP3,在这种命名法中E-LTP称为LTP1。[25]
诱导
后期LTP是由基因表达和蛋白质合成所引发的变化所诱导的,这个过程由E-LTP期间激活的蛋白激酶(如MAPK)的持续激发而引起。[25][26][31]事实上,MAPK,特别是细胞外信号调节激酶(ERK),可能是E-LTP和L-LTP之间的分子联系,因为许多E-LTP中涉及的信号通路,包括CaMKII和PKC,可以在ERK刺激下收敛。[31]最近的研究表明,L-LTP的诱导可以依靠重合的分子事件,即PKA激活和钙离子内流,来收敛CRTC1(TORC1)——cAMP反应元件结合蛋白(CREB)的一个强有力的转录激活体。LTP的协同性要求一种精确的分子符合,而且据推测,学习的过程也是如此。[32]
维持
激活后,ERK可以磷酸化一系列的细胞质和细胞核分子,最终导致蛋白质的合成以及L-LTP中观察到的形态学改变。[25]这些细胞质和细胞核分子包括CREB这样的转录因子。[26]ERK-介导的转录因子活性的变化可能会触发蛋白质的合成,为L-LTP的打下基础。一个这样的分子可能是蛋白激酶Mζ(PKMζ)——一种持续活跃激酶,它的合成能增强之后LTP的诱导。[33][34]PKMζ是非典型的PKC亚型,缺乏调节亚基,但尽管如此仍然能被构成地激活。[33]不同于其他激酶介导的LTP,PKMζ不只是活跃在LTP诱导后的第30分钟,而是后期LTP维持的必要条件。[33]因此,PKMζ对持久性记忆、而且很可能对维持长期记忆都是重要的。事实上,对大鼠海马体施用PKMζ抑制剂会导致逆行性失忆症,但短期记忆却是完整,这说明PKMζ对短期记忆没有作用。[34]最近的研究表明,PKMζ通过指挥突触支架蛋白质的运输和重组来维持L-LTP,[33][34]以为L-LTP的表达做准备。[33]然而更进一步的研究显示,缺乏PKMζ的转基因小鼠可以表现出正常的LTP,故而PKMζ的必要性尚有争论。[35]
表达
L-LTP中合成的蛋白质的种类只有少数是已知的。不管它们是何种蛋白,据认为它们有助于增加树突棘数目、比表面积、以及与L-LTP的表达相关联的神经递质的突触后敏感性。后者可能部分是由增强L-LTP过程中AMPA受体的合成而导致的。后期LTP也与突触前突出结合蛋白的合成和突触小泡数量增加有关,这表明L-LTP不仅诱导突触后细胞中蛋白质的合成,也对突触前细胞发生作用。像前面所提到的那样,突触后LTP诱导导致突触前蛋白质的合成,突触后和突触前细胞必然存在某种交流。这可能是通过逆行信使的合成而达成的。[25]
即使在仅限于突触后事件的研究中,科学家还没有确定的构成L-LTP基础的蛋白质合成的位置。具体来说,目前还不清楚蛋白质的合成到底发生在突触后胞体或树突中。尽管早在20世纪60年代就有人观察到树突中的核糖体(蛋白质合成机器的主要构件),但当时认为神经元中细胞体才是蛋白质合成的主要部位。这种判断直到20世纪80年代都没有受到严重挑战,据那时的研究者报道,观察到了树突中的蛋白质合成,而它们与细胞体的联系已被切断。[31]更近的研究表明,这种类型的树突蛋白质合成对某些类型的LTP是必需的。[36][37]
树突蛋白质合成的假说流行的原因之一是,它为LTP相关的特异性提供了一种可能的机理。具体而言,如果树突蛋白质合成确实是L-LTP的基础,只有接收LTP诱导刺激的树突棘会经历LTP,增强作用不会传播到相邻的突触。[31]相比之下,发生在胞体内的全域蛋白质合成,会使得蛋白质被运送到细胞的每一个区域,包括没有收到LTP诱导刺激的突触。树突蛋白质合成提供了一种机制的特殊性,全域蛋白质合成似乎就达不到这个要求。然而,突触标识假说却能够成功地统一解释全域蛋白质合成、突触的特异性和协同性。
逆行信号
逆行信号假说试图解释,LTP在突触后诱导和表达时,一些证据表明它在突触前同样有表达的现象。[16][28][38]因为正常的突触传递是有方向性的,从突触前传递到突触后细胞,因而这种传导信号被称为“逆行信号”。对于突触后发生、并部分在突触前表达的诱导,信息必须从发生表达的突触后细胞逆向传导给突触前的细胞。一旦有信息,将会启动级联事件,导致突触前成分的表达,如神经递质囊泡释放的概率增加。[39]
逆行信号目前是有争议的,一些研究者完全不相信突触前细胞参与LTP的表达。即使假说的支持者之间,信使的种类也存在争议。早期的研究者认为是一氧化氮,而最新的证据则表明很可能是细胞黏附蛋白。[16]
突触标识
树突蛋白质合成假说获得广泛接受之前,科学家普遍同意L-LTP相关蛋白质的合成发生在胞体。此外,还有认为这种合成的产品以一种非特异性的方式被运往细胞各个区域。因此,就必须解释蛋白质的合成发生在胞体内如何才能不损害LTP的输入特异性。突触标识假说试图解决这个难题,即胞体内合成蛋白质如何确保只达到已收到LTP诱导刺激的突触。
突触标识假说认为,已收到LTP诱导刺激的突触会合成一个“突触标记”,这个突触标记可能有助于捕捉可塑性相关的蛋白质,这些蛋白质从胞体运输到细胞各处。[40]对于裸鰓類生物加利福尼亚海兔LTP的研究显示,突触标记与LTP输入特异性有关。[41][42]一些证据表明,对于两个相距甚远的突触,一个突触LTP的诱导刺激驱动好几个信号级联,激发细胞核内的基因表达。同一个突触(而不是未刺激的突触)中,树突的蛋白质合成会建立一个短期的突触标记(不到三个小时)。基因表达的产物会运输到整个细胞,但只被有突触标记的突触捕获。因此,只有接收LTP的诱导刺激突触会表现出增强作用。
突触标识假说也可以解释LTP的关联性和协同性。关联性是指一个突触由于LTP而导致兴奋时,另一只受到微弱刺激的突触也会发生LTP。一般人们只想到强烈刺激的突触会历经LTP(仅微弱刺激不足以诱导其中任一突触的LTP),但实际上两个突触均会历经LTP。虽然微弱的刺激不能诱导蛋白质在胞体内合成的,它们可能会促进突触标识的合成。同时,对于单独通路的强烈刺激,能够诱导胞体内蛋白质的合成,可能促使生产可塑性相关的蛋白质,而运输到细胞各处。这样两个突触均表现出突触标识,故都将捕获蛋白质产物,致使经历强刺激和弱刺激的通路均表现出LTP。
协同性是指,两个单独施加时不足以诱导LTP的微弱刺激同时作用于两个突触时,两个突触均被激活。突触标识并不能解释多个微弱的刺激为何能形成协作,而足以刺激诱导LTP(这是前面描述的突触后兴奋叠加所能解释的)。相反,突触标识能够解释弱刺激突触的能力,其中任何一个不足以独立产生LTP,但却能够一起收到蛋白质合成产物。像之前描述的那样,这可能是通过微弱突触刺激后的本地突触标识的合成来完成的。
调制
| 调制物 | 目标 |
|---|---|
| β-肾上腺素受体 | cAMP、MAPK扩增 |
| 一氧化氮合酶 | 鸟苷酸环化酶、PKG、NMDAR |
| 多巴胺受体 | cAMP、MAPK扩增 |
| 代谢型谷氨酸受体 | PKC、MAPK扩增 |
如前所述,作为LTP基础的那些分子可被归类为介体或调制物。LTP介体是这样一种分子,如NMDA受体或钙,其存在和活动几乎是所有条件下激发LTP所必需的。而调制物则是可以改变LTP状态的一种分子,但对于它的产生或表达不是必需的。
除了上述的信号通路,海马体LTP可能会被多种调制物改变。例如,类固醇激素雌二醇可通过驱动CREB磷酸化和随后的树突棘生长来增强LTP。[43]此外,β-肾上腺素受体激动剂去甲肾上腺素可改变后期LTP所依赖的蛋白质的合成。[44]一氧化氮合酶活性也可能会影响随后鸟苷酸环化酶的激活和PKG。[45]同样,多巴胺受体的激活可通过cAMP/PKA信号通路增强LTP。[46][47]
与行为记忆的关联
虽然细胞培养中突触的LTP似乎为学习和记忆提供了一个良好的基础,LTP对于行为学习——即整个生物水平的学习的贡献,无法从体外研究中简单的推断出来。出于这个原因,科学家为探明LTP是否是一个活体动物学习和记忆的要求做了相当大的努力。LTP在恐惧情绪的建立上也起着至关重要的作用。
空间记忆

1986年,理查德·莫里斯提供了LTP的确是在体内形成记忆所必需的最早的一些证据。[48]他通过药理学方法修饰大鼠的海马体,以测试它们的空间记忆,因为海马体对空间学习的重要作用当时已然很明了。莫里斯对它们进行水迷宫训练,这些大鼠在漆黑的泳池内游泳,直到他们能够找到水面之下隐藏的平台。在这种训练中,正常大鼠会将隐蔽平台的位置与迷宫中布置的明显线索相关联。训练结束后,将一组大鼠的海马体用NMDA受体拮抗剂APV处理,而另一组则作为对照。然后两组均进行水迷宫空间记忆测试。对照组大鼠能够找到平台并从池中逃脱,而APV处理大鼠的表现明显不正常。此外,取自两组大鼠的海马体切片中,LTP本来很容易受控诱导,但在经APV处理的大鼠脑中却无法诱导。这提供了早期的证据,证明NMDA受体——乃至LTP——至少是某些类型的学习和记忆所必需的。
类似的,利根川进在1996年发现,海马体CA1区对活鼠空间记忆的形成至关重要。只有当大鼠处于特定区域——称为“位置区域”时,位于该CA1区的所谓位置细胞才会变得活跃。由于这些位置区域分布在整个环境中,一个种解释是,位置细胞组在海马体中能够构成地图。这些地图的精度决定了大鼠如何学习周围的环境,以及它如何探索环境。利根川发现破坏NMDA受体——特别是通过遗传学手段去除CA1区NR1亚基的大鼠,位置区域的产生明显没有对照组精确。也就是说,当NMDA受体受损时,大鼠会产生错误的空间地图。正如预期的那样,这些大鼠在空间测试中与对照组相比表现很差,进一步支持了LTP在空间学习中的作用。[49]
抑制性回避
2006年,乔纳森·惠特洛克和他的同事报道了一系列的实验,提供了关于LTP对行为记忆的作用的也许是最强有力的证据。[50]他们认为LTP是行为学习的基础,两个过程相互模仿相互禁锢。研究人员采用一种抑制性回避学习范式在明暗室两室装置中训练大鼠,大鼠进入暗室时会被施以足底电击。海马体CA1区的突触分析显示,抑制性回避训练诱导体内AMPA受体的磷酸化,与体外LTP时所观察到的类型相同,这说明抑制性回避训练能够模拟LTP。此外,训练中增强的突触不能在实验操作中进一步增强,否则会诱导LTP,也就是说,抑制性回避测试同时也能禁锢LTP。在回应文章中,蒂莫西·布利斯和同事表示,这些和相关实验“为LTP作为记忆的神经机制的情况下提供了实质的证明。”[51]
临床意义
LTP对疾病的作用比它在突触可塑性基本机制中的作用尚不太清楚。然而,LTP的改变可能会导致一些神经系统疾病,包括抑郁症、帕金森病、癫痫和神经性疼痛。[52]LTP受损可能对阿兹海默病和藥物成癮也有一定作用。
阿兹海默症
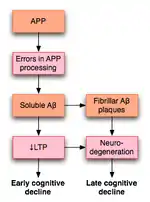
LTP引起了不少阿兹海默症(AD)研究者的注意。这是一种神经退行性疾病,可导致明显的认知能力下降和痴呆。这种恶性病变与发生在海马体和内侧颞叶结构的退行性改变有关。由于海马体对LTP的影响,一些人认为,认知能力下降可能与LTP受损有关。
2003年,在对已有文献进行考察的基础上,罗温等人提出一种模型,来解释LTP如何影响AD。AD很可能,至少部分的,由淀粉样前体蛋白(APP)处理过程的异常而导致。这种不正常的过程的结果是,这种蛋白的片段,称为β-淀粉样蛋白(Aβ)的积累。 Aβ既可以溶解形式,也可以纤维状形式存在。按照罗云的假说,APP处理异常导致可溶性Aβ的积累,会损害海马体LTP,并最终导致AD早期所表现出的认知能力下降的症状。[53]
AD也可能通过有别于Aβ的机制损害LTP。例如,研究表明,PKMζ酶积累于神经原纤维缠结,而神经原纤维缠结正是AD的病理标识。 PKMζ对于维持后期LTP至关重要。[54]
参考资料
- Paradiso, Michael A.; Bear, Mark F.; Connors, Barry W. . Hagerstwon, MD: Lippincott Williams & Wilkins. 2007: 718. ISBN 0-7817-6003-8.
- Cooke SF, Bliss TV. . Brain. 2006, 129 (Pt 7): 1659–73. PMID 16672292. doi:10.1093/brain/awl082.
- Bliss TV, Collingridge GL. . Nature. January 1993, 361 (6407): 31–39. PMID 8421494. doi:10.1038/361031a0.
- Williams RW, Herrup K. . Annu. Rev. Neurosci. 1988, 11 (1): 423–53 [2013-10-28]. PMID 3284447. doi:10.1146/annurev.ne.11.030188.002231. (原始内容存档于2018-01-03).
- Ramón y Cajal, Santiago. . Proceedings of the Royal Society of London. 1894, 55 (331-335): 444–468. doi:10.1098/rspl.1894.0063.
- Hebb, D. O. . New York: John Wiley. 1949. ISBN 0-471-36727-3.
- Ling GN, Gerard RW. . J Cell Comp Physiol. 1949, 34: 383–396.
- Terje Lømo. . Philos Trans R Soc Lond B Biol Sci. 2003, 358 (1432): 617–20. PMC 1693150. PMID 12740104. doi:10.1098/rstb.2002.1226.
- Lømo, Terje. . Acta Physiologica Scandinavica. 1966, 68 (Suppl 277): 128.
- Bliss T, Lømo T. . J Physiol. 1973, 232 (2): 331–56. PMC 1350458. PMID 4727084.
- Bliss T, Gardner-Medwin A. . J. Physiol. (Lond.). 1973, 232 (2): 357–74. PMC 1350459. PMID 4727085.
- Douglas R, Goddard G. . Brain Res. 1975, 86 (2): 205–15. PMID 163667. doi:10.1016/0006-8993(75)90697-6.
- Andersen P. . Philos. Trans. R. Soc. Lond., B, Biol. Sci. 2003, 358 (1432): 613–5. PMC 1693144. PMID 12740103. doi:10.1098/rstb.2002.1232.
- McEachern, JC; Shaw, CA. . Brain Research Review. June 1996, 22 (1): 51–92. PMID 8871785. doi:10.1016/0165-0173(96)00006-9. 8871785.
- Clugnet, MC; LeDoux JE. (PDF). J Neurosci. 1 August 1990, 10 (8): 2818–24. PMID 2388089.
- Malenka R, Bear M. . Neuron. 2004, 44 (1): 5–21. PMID 15450156. doi:10.1016/j.neuron.2004.09.012.
- Yasuda H, Barth A, Stellwagen D, Malenka R. . Nat Neurosci. 2003, 6 (1): 15–6. PMID 12469130. doi:10.1038/nn985.
- Harris E, Cotman C. . Neurosci Lett. 1986, 70 (1): 132–7. PMID 3022192. doi:10.1016/0304-3940(86)90451-9.
- Wigström H, Gustafsson B. . J. Physiol. (Paris). 1986, 81 (4): 228–36. PMID 2883309.
- Urban NN, Barrionuevo G. . J. Neurosci. July 1996, 16 (13): 4293–9 [2013-10-30]. PMID 8753890. (原始内容存档于2019-07-10).
- Kullmann DM, Lamsa K. . J. Physiol. (Lond.). March 2008, 586 (6): 1481–6. PMC 2375711. PMID 18187472. doi:10.1113/jphysiol.2007.148064.
- Malenka, Robert C. . Nature Reviews Neuroscience. 2003, 4 (11): 923–926. ISSN 1471-003X. doi:10.1038/nrn1258.
- McNaughton BL. . Philosophical transactions of the Royal Society of London. Series B, Biological sciences. April 2003, 358 (1432): 629–34. PMC 1693161. PMID 12740107. doi:10.1098/rstb.2002.1231.
- Abraham WC. . Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. April 2003, 358 (1432): 735–44. PMC 1693170. PMID 12740120. doi:10.1098/rstb.2002.1222.
- Lynch M. . Physiol Rev. 2004, 84 (1): 87–136 [2013-11-02]. PMID 14715912. doi:10.1152/physrev.00014.2003. (原始内容存档于2007-05-14).
- Sweatt J. . Learn Mem. 1999, 6 (5): 399–416. PMID 10541462. doi:10.1101/lm.6.5.399.
- Malinow R. . Philos Trans R Soc Lond B Biol Sci. 2003, 358 (1432): 707–14. PMC 1693162. PMID 12740116. doi:10.1098/rstb.2002.1233.
- Emptage N, Reid C, Fine A, Bliss T. . Neuron. 2003, 38 (5): 797–804. PMID 12797963. doi:10.1016/S0896-6273(03)00325-8.
- Frey U, Frey S, Schollmeier F, Krug M. . J Physiol. 490. 1 January 1996, (Pt 3) (Pt 3): 703–11. PMC 1158708. PMID 8683469.
- Frey U, Krug M, Reymann K, Matthies H. . Brain Res. 1988, 452 (1-2): 57–65. PMID 3401749. doi:10.1016/0006-8993(88)90008-X.
- Kelleher R, Govindarajan A, Tonegawa S. . Neuron. 2004, 44 (1): 59–73. PMID 15450160. doi:10.1016/j.neuron.2004.09.013.
- Kovács KA, Steullet P, Steinmann M, Do KQ, Magistretti PJ, Halfon O, Cardinaux JR. . PNAS. 2007, 104 (11): 4700–5. PMC 1838663. PMID 17360587. doi:10.1073/pnas.0607524104.
- Serrano P, Yao Y, Sacktor T. . J Neurosci. 2005, 25 (8): 1979–84. PMID 15728837. doi:10.1523/JNEUROSCI.5132-04.2005.
- Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton A, Sacktor T. . Science. 2006, 313 (5790): 1141–4. PMID 16931766. doi:10.1126/science.1128657.
- Volk, Lenora J.; Bachman, Julia L.; Johnson, Richard; Yu, Yilin; Huganir, Richard L. . Nature. 2 January 2013, 493 (7432): 420–423. doi:10.1038/nature11802.
- Kang H, Schuman E. . Science. 1996, 273 (5280): 1402–6. PMID 8703078. doi:10.1126/science.273.5280.1402.
- Steward O, Worley P. . Proc Natl Acad Sci USA. 2001, 98 (13): 7062–8. PMC 34623. PMID 11416188. doi:10.1073/pnas.131146398.
- Pavlidis P, Montgomery J, Madison D. . J Neurosci. 2000, 20 (12): 4497–505. PMID 10844019.
- Zakharenko S, Patterson S, Dragatsis I, Zeitlin S, Siegelbaum S, Kandel E, Morozov A. . Neuron. 2003, 39 (6): 975–90. PMID 12971897. doi:10.1016/S0896-6273(03)00543-9.
- Frey U, Morris R. . Nature. 1997, 385 (6616): 533–6. PMID 9020359. doi:10.1038/385533a0.
- Martin K, Casadio A, Zhu H, Yaping E, Rose J, Chen M, Bailey C, Kandel E. . Cell. 1997, 91 (7): 927–38. PMID 9428516. doi:10.1016/S0092-8674(00)80484-5.
- Casadio A, Martin K, Giustetto M, Zhu H, Chen M, Bartsch D, Bailey C, Kandel E. . Cell. 1999, 99 (2): 221–37. PMID 10535740. doi:10.1016/S0092-8674(00)81653-0.
- Segal M, Murphy D. . Neural Plast. 1999, 6 (3): 1–7. PMC 2565317. PMID 9920677. doi:10.1155/NP.1998.1.
- Straube T, Frey J. . Neuroscience. 2003, 119 (2): 473–9. PMID 12770561. doi:10.1016/S0306-4522(03)00151-9.
- Lu Y, Kandel E, Hawkins R. . J Neurosci. 1999, 19 (23): 10250–61. PMID 10575022.
- Frey U, Matthies H, Reymann K, Matthies H. . Neurosci Lett. 1991, 129 (1): 111–4. PMID 1833673. doi:10.1016/0304-3940(91)90732-9.
- Otmakhova N, Lisman J. . J Neurosci. 1996, 16 (23): 7478–86. PMID 8922403.
- Morris R, Anderson E, Lynch G, Baudry M. . Nature. 1986, 319 (6056): 774–6. PMID 2869411. doi:10.1038/319774a0.
- McHugh T, Blum K, Tsien J, Tonegawa S, Wilson M. . Cell. 1996, 87 (7): 1339–49. PMID 8980239. doi:10.1016/S0092-8674(00)81828-0.
- Whitlock J, Heynen A, Shuler M, Bear M. . Science. 2006, 313 (5790): 1093–7. PMID 16931756. doi:10.1126/science.1128134.
- Bliss T, Collingridge G, Laroche S. . Science. 2006, 313 (5790): 1058–9. PMID 16931746. doi:10.1126/science.1132538.
- Cooke SF, Bliss TV. . Brain: A Journal of Neurology. July 2006, 129 (Pt 7): 1659–73 [2013-11-08]. PMID 16672292. doi:10.1093/brain/awl082. (原始内容存档于2016-05-18).
- Rowan MJ, Klyubin I, Cullen WK, Anwyl R. . Philosophical transactions of the Royal Society of London. Series B, Biological sciences. April 2003, 358 (1432): 821–8. PMC 1693153. PMID 12740129. doi:10.1098/rstb.2002.1240.
- Crary JF, Shao CY, Mirra SS, Hernandez AI, Sacktor TC. . Journal of neuropathology and experimental neurology. April 2006, 65 (4): 319–26. PMID 16691113. doi:10.1097/01.jnen.0000218442.07664.04.
- Kauer JA, Malenka RC. . Nature reviews. Neuroscience. November 2007, 8 (11): 844–58. PMID 17948030. doi:10.1038/nrn2234.
- Wolf ME. . Molecular interventions. August 2003, 3 (5): 248–52. PMID 14993438. doi:10.1124/mi.3.5.248.
延伸阅读
- Bliss, T; Collingridge, G; Morris, R. . Oxford: Oxford University Press. 2004. ISBN 0-19-853030-7.
外部链接
- 研究者提供了学习机制的首个证据,PhysOrg.com2006年发布的Bear等人的研究报告。
- 关于杜奇鼠的短片 (RealPlayer格式)
- 《聪明的鼠》,Quantum ABC TV关于杜奇鼠的纪录片。
- MeSH(醫學主題詞)上面的Long-Term+Potentiation(美式英语)
