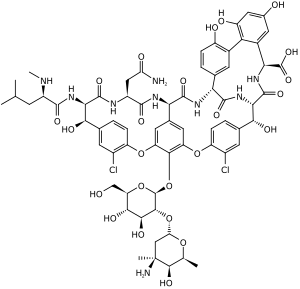
萬古黴素
萬古黴素(Vancomycin,INN)是一種糖肽類抗生素,用來治療許多细菌感染的抗细菌药抗生素[1]。治療皮膚感染、敗血症、心內膜炎、骨關節感染以及因耐甲氧西林金黄色葡萄球菌(MRSA)引起的脑膜炎時,通常建議採用靜脈注射作為第一線治療[2];而投藥時,劑量將依患者的血液濃度作調整。萬古黴素也是一種治療嚴重偽膜性結腸炎的口服藥,然而口服時藥效將會大為降低。[1],用來預防和治療革蘭氏陽性菌所造成的感染。傳統上,萬古黴素被用作「最後一線藥物」,用來治療所有抗生素均無效的嚴重感染。但由於越來越多的抗萬古黴素耐藥性病菌的出現,其地位漸漸被利奈唑胺和達托黴素所取代。
 | |
 | |
| 系统(IUPAC)命名名称 | |
|---|---|
未有 | |
| 临床数据 | |
| 妊娠分级 | |
| 给药途径 | 靜脈或口服 |
| 合法狀態 | |
| 合法状态 |
|
| 药代动力学数据 | |
| 生物利用度 | 可省略(口服) |
| 代谢 | 排泄時沒有改變 |
| 生物半衰期 | 4-11小時(成人) 6-10日(腎功能受損的成人) |
| 排泄 | 腎臟 |
| 识别 | |
| CAS注册号 | 1404-90-6 |
| ATC代码 | A07AA09 J01 |
| PubChem | CID 14969 |
| DrugBank | APRD01287 |
| 化学 | |
| 化学式 | C66H75Cl2N9O24 |
| 摩尔质量 | 1449.3 g·mol-1 |
常見的副作用包含注射至局部時的疼痛、过敏,有時候也可能引發聽力問題、低血壓或骨髓抑制等問題;對母乳餵養的患者似乎沒有問題,而對於懷孕患者的影響尚不明確,但目前沒有證據顯示會造成危害。[3]萬古黴素是一種糖肽類抗生素,藉由阻斷細胞壁生成來發揮作用。
萬古黴素於1954年上市[4],在最基本的健康照護系統中最必要的藥物清單世界卫生组织基本药物标准清单登錄有案[5],是一種常見藥物[3] ,一劑靜脈注射的批發價約在1.70-6美元左右[6];因為在美國靜脈注射劑型相當昂貴[1] ,靜脈注射的投藥方式可能會被取代[7]。萬古黴素目前由土壤細菌Amycolatopsis orientalis培養得來。
历史
万古霉素首先被EC Kornfeld(礼来公司员工)从一个传教士采集的婆罗洲丛林深处的土壤中分离出来。产生万古霉素的细菌最终被命名为东方拟无枝酸菌。[1]使用万古霉素的最初目的是治疗由耐青黴素的金黄色葡萄球菌引起的感染。[2][8]
最初,这种分离出来的化合物被简单的标记为05865,但是最后它有了一个通用的名称:Vancomycin(从单词 "vanquished" (征服)衍生而来)。[1]很快,万古霉素的优点就表现了出来:尽管在含万古霉素的培养基上经过多代的传代培养,葡萄球菌仍然未显示出明显的抗药性。当时,越来越多的葡萄球菌对青黴素类抗生素产生了抗药性,这使万古霉素很快在1958年获得了FDA的许可。Eli Lily公司首先生产并销售万古霉素,商标是 Vancocin。[2]
由于以下几个原因,万古霉素从来没有成为治疗金黄色葡萄球菌感染的一线药物:
- 万古霉素必须静脉注射给药,口服无法吸收。
- 抗β-内酰胺酶半合成青黴素类药物,如甲氧苯青黴素(以及其后继产品如乙氧萘(胺)青黴素,邻氯青黴素等)的快速发展,可以有效抑制对青黴素产生耐药性的细菌。
- 早期的实验中所用的纯度不高的万古霉素具有很强的耳毒性及肾毒性。[9]
这些发现导致万古霉素被降级为抗感染的“最后一线药物”。
2004年,礼来公司将Vancocin商标授权给美国的ViroPharma制药公司,英国的Flynn Pharma制药公司及澳大利亚的Aspen Pharmacare制药公司使用。万古霉素的专利在80年代早期就已经过期,但是FDA没有允许上述三家制药公司中任何一家生产的万古霉素在美国销售。
生物合成

万古霉素通过不同的非核糖体肽合成酶进行生物合成。[3]七个基团通过氨基酸相互连接,非核糖体肽合成酶决定了氨基酸的排列顺序。在万古霉素的基团通过氨基酸组装之前,氨基酸首先要经过修饰。L-酪氨酸被修饰为β-羟基,氯-酪氨酸和4-羟苯基甘氨酸残基。另一方面,醋酸根被用来合成3,5-二羟苯甘氨酸的苯环。[4]
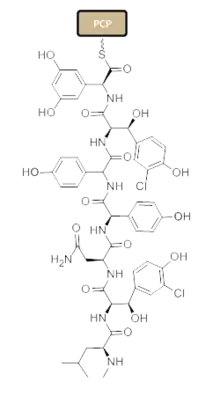
非核糖体肽合成酶在不同基团连接时发挥作用,通过在氨基酸之间形成肽键的方式延长肽链。[5]每个基团一般包括腺苷酰化结构域(A),肽载体蛋白结构域(PCP)以及肽键形成结构域(C)。在A结构域,特殊的氨基酸通过形成氨酰腺苷酶复合体的方式被激活,并与一个4`-磷酸泛酰巯基乙胺辅助因子通过硫酯化的方式结合。[6][7]这个复合物随后被AMP运送至PCP。PCP利用结合的4'-磷酸泛酰巯基乙胺辅基来运送肽链和它们的前体。[10]图片1显示了生物合成万古霉素所必需的基团。在万古霉素的生物合成中,还存在有其他修饰结构域如差向异构结构域(E),用来将某些氨基酸进行立体异构;硫酯酶结构域(TE)用来催化环化反应以及通过断裂硫酯键从而去除某些不需要的分子。

一种多酶复合体(包括肽合成酶CepA, CepB, 和 CepC)负责组装构成万古霉素的七肽结构。CepA, CepB, 和 CepC的结构与其他的肽合成酶(如表面活性肽合成酶以及短杆菌肽合成酶)非常相似。每个肽合成酶把不同的氨基酸按一定的顺序编码从而激活不同的结构域。CepA编码基团1,2和3,CepB编码基团4,5和6,CepC编码基团7。这三个肽合成酶的基因序列位于细菌染色体的起始段,长度为27kb。[5]
在线性的七肽分子合成之后,万古霉素还要进一步的进行翻译后的修饰,在一些酶的作用下进行氧化交联和糖基化,反式化反应等,以产生生物活性。八种酶,即开放阅读框架(ORF)7,8,9,10,11,14,18,20和21被用来转换这种链状七肽。ORF 7,8,9和20是P450酶,ORF10和18是不含血红素的卤过氧化物酶。ORF9和14被认为可能是羟化酶。[11]在这些酶的帮助下,β-羟基被引入到酪氨酸的2号和6号位,连接发生在5号环与7号环,4号环与6号环,以及4号环与2号环之间。另外,一种过氧化物酶将一个氯原子通过氧化反应连接到2号环与6号环上。[5]
药理学与结构
万古霉素是三环糖苷化非核糖体肽的一种分支产物,由放线菌属的东方拟无枝酸菌(以前被命名为东方诺卡菌)通过发酵产生。
万古霉素的作用机制是,通过抑制革兰氏阳性菌的细胞壁的合成而使细菌无法生存。这种机制以及革兰氏阴性菌的生理特征意味着,万古霉素对于革兰氏阴性菌几乎无效(除了一些非淋菌性的奈瑟菌属细菌)。
特别的是,万古霉素阻止N-乙酰胞壁酰基(NAM-)和n-乙酰葡糖酰基(NAG-)参与肽聚糖骨架的形成,而肽聚糖骨架是革兰氏阳性菌细胞壁的主要成分。
万古霉素中大量的亲水基团可以形成氢键,与NAM-肽和NAG-肽中D-丙氨酰丙氨酸末端的部分相互作用。通常这是一种有5个作用点的反应。这样万古霉素分子就被氢键“捆绑”在了D-丙氨酰丙氨酸上,阻止了NAM-肽与NAG-肽参与肽聚糖骨架的形成。
万古霉素存在阻转异构现象。由于氯酪氨酸残基旋转特性的限制,万古霉素有两种典型的旋转异构体。此种构象异构的药物具有更好的热力学稳定性,以及更高的药效。
临床应用
适应症
万古霉素在临床上被用于治疗由对其他抗生素不敏感的革兰氏阳性菌所引起的严重的致命性感染。但是,万古霉素不应该被用于治疗对甲氧西林(甲氧苯青黴素)敏感的金黄色葡萄球菌引起的感染,因为其疗效较萘夫西林(乙氧萘(胺)青黴素)类青黴素弱。[12][13]
由于越来越多耐万古霉素的肠球菌菌群的出现,美国疾病控制中心所属的控制院内感染顾问委员会对临床万古霉素的应用提出了一些指导方针。根据这个指导方针,仅在如下情况下谨慎的使用万古霉素:[14]
- 由高致病性抗青黴素类细菌(耐甲氧西林金黄色葡萄球菌和多抗性表皮葡萄球菌)所引起的感染,或者治疗对青黴素高度过敏的个别病例。
- 治疗经甲硝唑治疗无效或治疗后复发的假膜性结肠炎。
- 治疗对β-内酰胺类抗生素严重过敏的革兰氏阳性菌感染的病例。
- 用于对青黴素高度过敏的个体进行感染性心内膜炎的抗菌性预防。
- 预防在耐甲氧西林金黄色葡萄球菌和多抗性表皮葡萄球菌分布广泛的医疗机构进行有假体植入的外科手术时可能引发的感染。
不良反应
尽管为了降低不良反应的发生几率,万古霉素的血药浓度常常会受到监控,但是这种监控的价值仍然有争议。[15]血药峰浓度和最低有效浓度是最常被监控的項目。此外为了临床研究,药-时曲线(反应血药浓度随时间变化的曲线)以下的区域的浓度也常常受到监测。在血药最低有效浓度时观察毒性反应是最好的选择。[16]
万古霉素常见的不良反应(在≥1%的病例中发生)有:注射区域(剧烈)疼痛、血栓静脉炎。
肾毒性和耳毒性是早期生产的不纯的万古霉素产生的副作用,在50年代中期进行的万古霉素临床试验中显得尤其严重。[17][18]然而在后来的试验中,由于采用更纯净的万古霉素,肾毒性的发生率变得很低(0.1–1%的病例中发生)。但这个结论强调,试验中万古霉素未与氨基糖苷类抗生素合用。[19]
罕见的不良反应(<0.1%的病例中发生)包括:过敏反应、中毒性表皮溶解坏死、多形性红斑、红人综合症(见下文)、二次感染、血小板下降、嗜中性粒细胞减少症、白细胞减少症、耳鸣、眩晕、耳毒性(见下文)。[14]
最近的研究强调,万古霉素可以引发患者体内产生抗血小板抗体,引起严重的血小板减少症以及由皮下红斑、瘀斑和紫癜引起的出血。[20]
静脉给药和口服给药
在治疗全身性感染时,万古霉素需要通过静脉给药,因为万古霉素不能被肠道吸收。作为一个具有亲水性的大分子,万古霉素很难穿过肠壁的绒毛细胞细胞膜。唯一需要口服给药的适应症是假膜性肠炎,因为必须经过口服药物才能到达感染部位。在口服给药的情况下,排泄物中的万古霉素浓度在500 µg/ml左右。[21]
通过雾化器吸入万古霉素进行治疗有时也会被采用(非规范性治疗方法),以治療由各种原因引起的上呼吸道及下呼吸道感染。万古霉素的腐蚀性也使用中央静脉导管给药时容易发生血栓静脉炎。[22]
红人综合症
静脉给药时,万古霉素必须在溶剂稀释的条件下缓慢给药,最短给药时间为60分钟(一次总给药量大于500mg时最大给药速度小于10 mg/min)。[14]这是因为静脉给药时局部疼痛和血栓静脉炎的发生率很高,以及为了避免一些输液反应如红人综合症(或称红脖综合症)的发生。红人综合症通常发生在开始输液后4-10分钟,或刚刚输液完成后,通常表现为面部、颈部以及上肢躯干部潮红或出現红色皮疹。这些症状是由于非特异性肥大细胞脱颗粒所引起而并非由IgE介导的变态反应。抗组胺药物如苯海拉明可以治疗或者预防这些症状的发生。缓慢输液也可以减少发生的机率。[23][24]
治疗药物监测
万古霉素的疗效被认为是与时间相关联的,这意味着血药浓度高于其最低抑菌浓度的持续时间决定了它的抗菌效果。因此,血药峰浓度并未显示出与其毒性或抗菌效果有关联。所以大多数情况下精确的血药浓度监测并不需要。仅在下列情况下需要进行治疗药物监测:
- 病人同时使用氨基糖苷类抗生素
- 病人的药代动力学参数(可能)改变
- 病人在治疗期间需要进行血液透析
- 短期大剂量或长期使用万古霉素治疗
- 病人有肾功能不全
在这些情况下,需要测量血药最低浓度。[14][25][26][27]
近年来学界改变了治疗时万古霉素血药浓度水平的标准。早期的学者认为治疗期间,万古霉素的血药峰浓度应该在30-40mg/L,最低浓度在5-10mg/L。[28]最新的建议为,在用于治疗炎症感染是,不需要限制血药峰浓度,最低浓度应该在15-20mg/L。[29]
毒性
传统上认为,万古霉素具有很强的肾毒性及耳毒性。早期调查中发现一些听力及肾功能受损的病人血清中万古霉素的水平很高,随后一些医学参考文献刊登了这些调查报告。然而,自从70年代万古霉素被大量应用于治疗耐甲氧西林金黄色葡萄球菌以后,临床上发现先前报告的严重的毒性并没有大范围出现。这被归结于现在使用的万古霉素中已经移除了早期的不纯净的成分,尽管未对这些不纯净的成分进行过毒性试验。[8]
肾毒性
随后,有学者对大量报告万古霉素相关的肾毒性病例进行了重新的调查。调查发现大部分病例同时使用了其他已知有肾毒性的药物,尤其是氨基糖苷类抗生素。其余的病例大多数有其他的干扰因素或者数据不完整,不足以证明万古霉素与肾功能损害有一定的关系。
1994年,坎图和他的同事发现,在文献记载的82宗发生肾毒性的病例中,仅有3宗是单纯使用万古霉素治疗的。[25]一些试图估计万古霉素相关性肾功能损害发生几率的前瞻性研究及回顾性分析大多在方法上有缺陷,因而产生许多不同的结果。公认最好方法得出的结论是万古霉素相关性肾功能损害的发生几率在5%~7%之间。这和两种公认的无肾毒性的抗生素,头孢孟多和苄青霉素所引起肾功能损伤的几率几乎相等。
此外,万古霉素的血清浓度和肾毒性发生几率是否相关,目前仍没有一致的结论。有些研究指出在万古霉素血药谷浓度超过10 µg/mL时,肾毒性的发生几率增加。但是其他的重复性试验并未能取得相同的结果。在治疗安全的范围内,万古霉素仍有引起肾毒性的报告。总之,万古霉素引起肾毒性的几率被高估了。而且,没有证据显示,当肾毒性发生时,改变万古霉素的血药浓度能阻止肾毒性的进一步加重。
耳毒性
确定萬古霉素引起耳毒性的几率更加困难,因为缺少有说服力的病例。普遍的观点是,万古霉素极少引起耳毒性症状。 万古霉素血药浓度与耳毒性的相关性也是不确定的。有的病例在万古霉素血清浓度超过 80 µg/mL时出現耳毒性症状,可是同样有在治疗安全范围的血清浓度下产生耳毒性病例的报告。因此,不能证明使用治疗药物检测的方法保持治疗安全范围的血药浓度可以防止耳毒性症状的发生。
与其他有肾毒性药物的相互作用
对于万古霉素研究的另一个疑问是,万古霉素能否加强其他有肾毒性药物的毒性,如果能的话,能够加强多少呢?临床研究得出许多不同的结论。但是动物模型显示,当万古霉素与其他的肾毒性药物如氨基糖苷类抗生素合并使用时,肾毒性会加强。但是,加强的程度仍然没有被证实与药物使用剂量与血清浓度有相关性。
抗生素耐药性
内在性耐药
有一些革兰氏阳性菌被发现对万古霉素有内在性耐药。这些菌种属于明串珠菌属和小球菌属,但是这些细菌极少感染人类。[30]大多数乳酸杆菌属菌种对万古霉素也有内在性抗性[30](除了一些(但不是全部)的嗜乳酸杆菌属的菌株[31])。
大多数革兰氏阴性菌对万古霉素具有内在性抗性,因为大分子的糖肽不能渗透进革兰氏阴性菌的外膜。[32](除了一些非淋菌性奈瑟菌属菌种)[33]
获得性耐药
获得性抗万古霉素抗性是一个越来越严重的问题,尤其是对于像医院这样的医疗机构来说。作为抗革兰氏阳性菌感染的“最后一道防线”,越来越多的抗万古霉素细菌将导致普遍性的致命性细菌感染。抗万古霉素肠球菌在1987年被发现,万古霉素抗性在90年代以及2000年后出现于常见致病微生物中,包括中介度耐万古霉素金黄色葡萄球菌以及抗万古霉素金黄色葡萄球菌和抗万古霉素艰难梭菌。[34][35]有些人怀疑,应用于农业领域的另一种糖肽类抗生素阿伏霉素是导致抗万古霉素菌种出现的原因之一。
对万古霉素产生抗性的机制之一是肽聚糖骨架中NAM-肽和NAG-肽末端的氨基酸残基发生改变。一般末端的氨基酸为D-丙氨酰丙氨酸,是与万古霉素连接的部分。如果末端氨基酸残基发生改变,如变成D-丙氨酰乳酸或D-丙氨酰丝氨酸,那么万古霉素和NAM-肽或NAG-肽之间只有4个氢键发生相互作用。这一个氢键的缺失使二者的亲和力下降了1,000倍。
在肠球菌属这个氨基酸残基改变的原因是相关酶的表达。在肠球菌属和粪肠球菌属,3个主要的耐药性变种的特征已经被详细的描述出来。
- 变种A- 对万古霉素及替考拉宁有耐药性,在此两种药物的作用下被诱导分化出来。
- 变种B- 耐药性水平低,由万古霉素诱导分化,但是仍然对替考拉宁敏感
- 变种C- 临床重要性最低,仅对万古霉素有基本的耐药性。
新的抗生素如利奈唑胺和达托霉素的发展和应用预计将会延迟,但是在不久的将来,对所有已知抗生素均有耐受性的菌种一定会出现。
参考资料
- Levine DP. . Clin Infect Dis. 2006, 42 (Suppl 1): S5–12. doi:10.1086/491709.
- Moellering, RC Jr. . Clin Infect Dis. 2006, 42: S3–S4. PMID 16323117.
- Samel SA, Marahiel MA, Essen LO. . Mol Biosyst. May 2008, 4 (5): 387–93. PMID 18414736. doi:10.1039/b717538h.
- Dewick, Paul M. . New York: Wiley. 2002. ISBN 978-0-471-49641-0.
- van Wageningen AM, Kirkpatrick PN, Williams DH; 等. . Chem. Biol. March 1998, 5 (3): 155–62. PMID 9545426. doi:10.1016/S1074-5521(98)90060-6.
- Schlumbohm W, Stein T, Ullrich C; 等. . J. Biol. Chem. December 1991, 266 (34): 23135–41. PMID 1744112.
- Stein T, Vater J, Kruft V; 等. . J. Biol. Chem. June 1996, 271 (26): 15428–35. PMID 8663196. doi:10.1074/jbc.271.26.15428.
- Donald P. . Clin Infect Dis. 2006, 42: S5–S12. PMID 16323120. doi:10.1086/491709.
- Griffith RS. . Rev Infect Dis. 1981, 3: S2004.
- Kohli RM, Walsh CT, Burkart MD. . Nature. August 2002, 418 (6898): 658–61. PMID 12167866. doi:10.1038/nature00907.
- Solenberg PJ, Matsushima P, Stack DR, Wilkie SC, Thompson RC, Baltz RH. . Chem. Biol. March 1997, 4 (3): 195–202. PMID 9115410. doi:10.1016/S1074-5521(97)90288-X.
- Small PM, Chambers HF. . Antimicrob Agents Chemother. 1990, 34: 1227–31. PMID 2393284.
- Gonzalez C, Rubio M, Romero-Vivas J, Gonzalez M, Picazo JJ. . Clin Infect Dis. 1999, 29: 1171–7. PMID 10524959. doi:10.1086/313440.
- Rossi S, editor. Australian Medicines Handbook 2006. Adelaide: Australian Medicines Handbook; 2006. ISBN 978-0-9757919-2-9
- Cantú TG, Yamanaka-Yuen NA, Lietman PS. . Clin Infect Dis. 1994, 18 (4): 533–43. PMID 8038306.
- Lodise TP, Patel N, Lomaestro BM, Rodvold KA, Drusano GL. . Clin Infect Dis. 2009, 49 (4): 507–514. doi:10.1086/600884.
- Moellering RC Jr. . Clin Infect Dis. 2006, 42 (Suppl 1): S3–4. PMID 16323117.
- Levine DP. . Clin Infect Dis. 2006, 42 (Suppl 1): S5–12. PMID 16323120.
- Farber BF, Moellering RC Jr. . Antimicrob Agents Chemother. 1983, 23: 138.
- Drygalski A, Curtis BR. . N Engl J Med. 2007, 356: 904. PMID 17329697. doi:10.1056/NEJMoa065066.
- Edlund C, Barkholt L, Olsson-Liljequist B, Nord CE. . Clin Infect Dis. 1997, 25: 729–32.
- . [2009-10-29]. (原始内容存档于2011-03-20).
- Sivagnanam S, Deleu D. Red man syndrome. Crit Care 2003;7(2):119–120. PMID 12720556. (full text 的存檔,存档日期2006-05-08.)
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. ISBN 978-0-7216-2921-6.
- Cantu TG, Yamanaka-Yuen NA, Lietman PS. Serum vancomycin concentrations: reappraisal of their clinical value. Clin Infect Dis 1994;19(6):1180-2. PMID 8038306
- Moellering RC Jr. Monitoring serum vancomycin levels: climbing the mountain because it is there? Clin Infect Dis 1994;18(4):544-6. PMID 8038307
- Karam CM, McKinnon PS, Neuhauser MM, Rybak MJ. Outcome assessment of minimizing vancomycin monitoring and dosing adjustments. Pharmacotherapy 1999;19(3):257-66. PMID 10221365
- Geraci J. . Mayo Clin Proc. 1977, 52: 631–4. PMID 909314.
- Rybak M, Lomaestro B, Rotschafer JC; 等. . American Journal of Health-System Pharmacy. 2009, 66 (1): 82–98. doi:10.2146/ajhp080434.
- Swenson JM, Facklam RR, Thornsberry C. . Antimicrob Agents Chemother. 1990, 34: 543–49.
- Hamilton-Miller JM, Shah S. . Lett Appl Microbiol. 1998, 26: 153–54. doi:10.1046/j.1472-765X.1998.00297.x.
- Quintiliani R Jr, Courvalin P. . Murray PR, Baron EJ, Pfaller MA, Tenover FC, Yolken RH (编). 6th. Washington DC: ASM Press. 1995: 1319. ISBN 978-1-55581-086-3.
- Geraci JE, Wilson WR. . Rev Infect Dis. 1981,. 3(Suppl): S250–58.
- Smith TL, Pearson ML, Wilcox KR, Cruz C, Lancaster MV, Robinson-Dunn B, et al. Emergence of vancomycin resistance in Staphylococcus aureus. Glycopeptide-Intermediate Staphylococcus aureus Working Group. N Engl J Med 1999;340(7):493-501. PMID 10021469
- McDonald LC, Killgore GE, Thompson A, et al. Emergence of an epidemic, toxin gene variant strain of Clostridium difficile responsible for outbreaks in the United States between 2000 and 2004. N Engl J Med 2005;353:2433-2441. PMID 16322603
