血管性水肿
血管神经性水肿是真皮、皮下组织、黏膜的局部肿胀。[1][3]可发生于面部、舌头、喉、腹部、四肢。[1]常与荨麻疹相关,荨麻疹是皮肤的红肿。[1][3] Onset is typically over minutes to hours.[1]
| 血管神经性水肿 | |
|---|---|
| 同义词 | 血管性水肿,Angiooedema, Quincke's edema, angioneurotic edema |
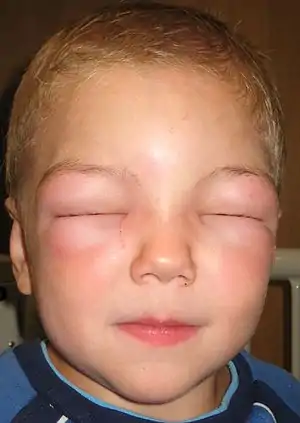 | |
| 血管神经性水肿: 患儿由于眼皮肿胀不能睁眼. | |
| 症状 | 局部肿胀 [1] |
| 常見始發於 | 几分钟到几小时[1] |
| 类型 | 组胺调节, 缓激肽调节[1] |
| 风险因子 | 家族史[2] |
| 診斷方法 | 对症治疗[2] |
| 相似疾病或共病 | 全身型過敏性反應, 膿瘍, 接触性皮炎[2] |
| 治療 | 插管, 环甲状软骨切开术[1] |
| 藥物 | 组胺: 抗組織胺藥, 皮質類固醇, 肾上腺素[1] 缓激肽: C1酯酶抑制物, 艾卡拉肽, 艾替班特, 新鲜冷冻血浆[1] |
| 盛行率 | ~100,000每年(美国)[1] |
| 分类和外部资源 | |
| 醫學專科 | 免疫学 |
| ICD-9-CM | 995.1 |
| OMIM | 106100、610618、106100、610618 |
| DiseasesDB | 13606 |
| MedlinePlus | 000846 |
| eMedicine | 756261、135208、885100 |
| Patient UK | 血管神经性水肿 |
基本机制涉及组胺或缓激肽。[1] 与组胺相关的是由于对过敏原的过敏反应,如蚊虫叮咬,食物,或药品。[1] 与缓激肽相关的是遗传问题称作获得性C1酯酶抑制剂缺乏,药物有血管紧张素转换酶抑制剂, 或淋巴组织增生性疾病.[1]
为保护呼吸道通畅,对呼吸道特别是喉部发作水肿,必要时应进行气管插管或环甲状软骨切开术。[1]组胺相关血管神经性水肿可抗組織胺藥:对症治疗常采用抗组胺受体H1拮抗剂,对顽固的、应用抗组胺受体拮抗剂无效的患者,可合并应用抗组胺受体H2拮抗剂如西咪替丁(甲氰咪呱)或兰替丁,有时可取得满意效果。酮体芬亦可合并使用。拟交感神经药物主要用于急性荨麻疹和(或)神经性水肿,尤其是喉水肿患者,应用0.1%肾上腺素皮下注射,对严重急性过敏性反应可隔20~30分钟注射。同时给予糖皮質類固醇激素静脉滴注,氨茶碱口服或静脉注射。[1] 缓激肽相关的疾病可用C1酯酶抑制物, 艾卡拉肽, 艾替班特治疗。[1] Fresh frozen plasma may be used instead.[1] In the United States the disease affects about 100,000 people a year.[1]
症状
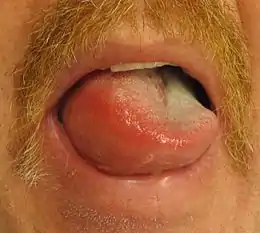

The skin of the face, normally around the mouth, and the mucosa of the mouth and/or throat, as well as the tongue, swell over the period of minutes to hours. The swelling can also occur elsewhere, typically in the hands. The swelling can be itchy or painful. There may also be slightly decreased sensation in the affected areas due to compression of the nerves. Urticaria (hives) may develop simultaneously.
In severe cases, stridor of the airway occurs, with gasping or wheezy inspiratory breath sounds and decreasing oxygen levels. Tracheal intubation is required in these situations to prevent respiratory arrest and risk of death.
Sometimes, the cause is recent exposure to an allergen (e.g. peanuts), but more often it is either idiopathic (unknown) or only weakly correlated to allergen exposure.
In hereditary angioedema, often no direct cause is identifiable, although mild trauma, including dental work and other stimuli, can cause attacks.[4] There is usually no associated itch or urticaria, as it is not an allergic response. Patients with HAE can also have recurrent episodes (often called "attacks") of abdominal pain, usually accompanied by intense vomiting, weakness, and in some cases, watery diarrhea, and an unraised, nonitchy splotchy/swirly rash. These stomach attacks can last one to five days on average, and can require hospitalization for aggressive pain management and hydration. Abdominal attacks have also been known to cause a significant increase in the patient's white blood cell count, usually in the vicinity of 13,000 to 30,000. As the symptoms begin to diminish, the white count slowly begins to decrease, returning to normal when the attack subsides. As the symptoms and diagnostic tests are almost indistinguishable from an acute abdomen (e.g. perforated appendicitis) it is possible for undiagnosed HAE patients to undergo laparotomy (operations on the abdomen) or laparoscopy (keyhole surgery) that turns out to have been unnecessary.
HAE may also cause swelling in a variety of other locations, most commonly the limbs, genitals, neck, throat and face. The pain associated with these swellings varies from mildly uncomfortable to agonizing pain, depending on its location and severity. Predicting where and when the next episode of edema will occur is impossible. Most patients have an average of one episode per month, but there are also patients who have weekly episodes or only one or two episodes per year. The triggers can vary and include infections, minor injuries, mechanical irritation, operations or stress. In most cases, edema develops over a period of 12–36 hours and then subsides within 2–5 days.
诊断
The diagnosis is made on the clinical picture. Routine blood tests (complete blood count, electrolytes, renal function, liver enzymes) are typically performed. Mast cell tryptase levels may be elevated if the attack was due to an acute allergic (anaphylactic) reaction. When the patient has been stabilized, particular investigations may clarify the exact cause; complement levels, especially depletion of complement factors 2 and 4, may indicate deficiency of C1-inhibitor. HAE type III is a diagnosis of exclusion consisting of observed angioedema along with normal C1 levels and function.
The hereditary form (HAE) often goes undetected for a long time, as its symptoms resemble those of more common disorders, such as allergy or intestinal colic. An important clue is the failure of hereditary angioedema to respond to antihistamines or steroids, a characteristic that distinguishes it from allergic reactions. It is particularly difficult to diagnose HAE in patients whose episodes are confined to the gastrointestinal tract. Besides a family history of the disease, only a laboratory analysis can provide final confirmation. In this analysis, it is usually a reduced complement factor C4, rather than the C1-INH deficiency itself, that is detected. The former is used during the reaction cascade in the complement system of immune defense, which is permanently overactive due to the lack of regulation by C1-INH.
Angioedema is classified as either hereditary or acquired.
获得性血管性水肿
Acquired angioedema (AAE) can be immunologic, nonimmunologic, or idiopathic.[5] It is usually caused by allergy and occurs together with other allergic symptoms and urticaria. It can also occur as a side effect to certain medications, particularly ACE inhibitors. It is characterized by repetitive episodes of swelling, frequently of the face, lips, tongue, limbs, and genitals. Edema of the gastrointestinal mucosa typically leads to severe abdominal pain; in the upper respiratory tract, it can be life-threatening.[6]
遗传型血管性水肿
Hereditary angioedema (HAE) exists in three forms, all of which are caused by a genetic mutation inherited in an autosomal dominant form. They are distinguished by the underlying genetic abnormality. Types I and II are caused by mutations in the SERPING1 gene, which result in either diminished levels of the C1-inhibitor protein (type I HAE) or dysfunctional forms of the same protein (type II HAE). Type III HAE has been linked with mutations in the F12 gene, which encodes the coagulation protein factor XII. All forms of HAE lead to abnormal activation of the complement system, and all forms can cause swelling elsewhere in the body, such as the digestive tract. If HAE involves the larynx, it can cause life-threatening asphyxiation.[7] The pathogenesis of this disorder is suspected to be related to unopposed activation of the contact pathway by the initial generation of kallikrein and/or clotting factor XII by damaged endothelial cells. The end product of this cascade, bradykinin, is produced in large amounts and is believed to be the predominant mediator leading to increased vascular permeability and vasodilation that induces typical angioedema "attacks".[8]
病理学
缓激肽在各种血管性水肿中扮演了关键角色。[9] 这种肽是强力血管舒張并增加血管通透,导致组织液快速累积。这在脸部特别显著,因为脸部皮肤相对缺少结缔组织支撑,容易形成水肿。多种细胞类型在刺激下会释放缓激肽。不同的干涉缓激肽生产或讲解的机制能导致血管性水肿。ACE inhibitors block 血管紧张素转化酶(ACE)能降解缓激肽。遗传性血管性水肿是导致缓激肽生成的补体系统持续活化,因为缺乏相应的抑制剂:C1酯酶(C1INH)。这种丝氨酸蛋白酶抑制剂能抑制C1r、C1s与C1q的关联,阻止了C1复合体的生成,从而激活了其他的补体系统的蛋白。此外还抑制了相应的一些凝血蛋白,虽然对出血与血栓形成的干扰效果是有限的。
有三类遗传性血管性水肿
- 第一类 - C1INH的低浓度 (85%);
- 第二类 - C1INH浓度正常,但缺乏功能 (15%);
- 第三类 - C1INH无异常, 伴性遗传主要影响女性; pregnancy与使用甾体激素者会加重 (发生频率不定).[10] 与哈格曼因子基因变异有关。[11]
血管性水肿也可能是生成了C1INH抗体,这是一种自體免疫性疾病。与淋巴瘤的发展有关。
食物消化吸收后可能带来自己的血管扩张剂,如酒精饮料或桂皮,可加重病情。菠萝蛋白酶与薑黃合用能减轻症状。[12]中药分别称两种效果为“发货”与抗炎消肿。
治疗
过敏
In allergic angioedema, avoidance of the allergen and use of antihistamines may prevent future attacks. Cetirizine is a commonly prescribed antihistamine for angioedema. Some patients have reported success with the combination of a nightly low dose of cetirizine to moderate the frequency and severity of attacks, followed by a much higher dose when an attack does appear. Severe angioedema cases may require desensitization to the putative allergen, as mortality can occur. Chronic cases require steroid therapy, which generally leads to a good response. In cases where allergic attack is progressing towards airway obstruction, epinephrine may be life-saving.
药物导致
ACE inhibitors can induce angioedema.[13][14][15] ACE inhibitors block the enzyme ACE so it can no longer degrade bradykinin; thus, bradykinin accumulates and causes angioedema.[13][14] This complication appears more common in African-Americans.[16] In people with ACE inhibitor angioedema, the drug needs to be discontinued and an alternative treatment needs to be found, such as an angiotensin II receptor blocker (ARB)[17] which has a similar mechanism but does not affect bradykinin. However, this is controversial, as small studies have shown some patients with ACE inhibitor angioedema can develop it with ARBs, as well.[18][19]
遗传性
In hereditary angioedema, specific stimuli that have previously led to attacks may need to be avoided in the future. It does not respond to antihistamines, corticosteroids, or epinephrine. Acute treatment consists of C1-INH (C1-esterase inhibitor) concentrate from donor blood, which must be administered intravenously. In an emergency, fresh frozen blood plasma, which also contains C1-INH, can also be used. However, in most European countries, C1-INH concentrate is only available to patients who are participating in special programmes. The medications ecallantide and icatibant may be used to treat attacks.[1] In 2017 these medications cost between 5,700 and 14,000 美元 per dose in the United States, prices that tripled in two years.[20]
Future attacks of hereditary angioedema can be prevented by the use of androgens such as danazol, oxandrolone or methyltestosterone. These agents increase the level of aminopeptidase P, an enzyme that inactivates kinins;[21] kinins (especially bradykinin) are responsible for the manifestations of angioedema. 活性减弱的雄性激素如达那唑、司坦唑(康力龙)、羟甲烯龙(康复龙)等治疗先天性C1INH缺陷,可纠正其生化缺损并有预防发作的效用,但不能用于小儿和孕妇。
历史
1882年,Heinrich Quincke首次临床报告此病。[23] 虽然有些更早的临床描述。[24][25][26]
1888年,William Osler认为某些是由于遗传导致的,称“遗传性血管神经性水肿”。[27]
1963年证实了C1酯酶抑制剂匮乏是病因。[28]
参见
- Drug-induced angioedema
- Gleich's syndrome (unexplained angioedema with high eosinophil counts)
参考文献
- Bernstein, JA; Cremonesi, P; Hoffmann, TK; Hollingsworth, J. . International journal of emergency medicine. December 2017, 10 (1): 15. PMID 28405953.
- Caterino, Jeffrey M.; Kahan, Scott. . Lippincott Williams & Wilkins. 2003: 133. ISBN 9781405103572. (原始内容存档于2017-09-10) (英语).
- Habif, Thomas P. 5. Elsevier Health Sciences. 2009: 182. ISBN 0323080375. (原始内容存档于2017-09-10) (英语).
- Bork K; Barnstedt Se. . J Am Dent Assoc. August 2003, 134 (8): 1088–94. PMID 12956349. doi:10.14219/jada.archive.2003.0323. (原始内容存档于2012-07-23).
- Axelrod, S; Davis-Lorton, M. . The Mount Sinai journal of medicine, New York. 2011, 78 (5): 784–802. PMID 21913206. doi:10.1002/msj.20288.
- Moon, MD, Amanda T.; Heymann, MD, Warren R. . MedScape. [1 October 2015]. (原始内容存档于5 September 2015).
- Zuraw B.L. . N. Engl. J. Med. September 2008, 359 (10): 1027–36. PMID 18768946. doi:10.1056/NEJMcp0803977.
- Loew, Burr. . MedScape. [19 October 2012]. (原始内容存档于22 October 2012).
- Bas M, Adams V, Suvorava T, Niehues T, Hoffmann TK, Kojda G. . Allergy. 2007, 62 (8): 842–56. PMID 17620062. doi:10.1111/j.1398-9995.2007.01427.x.
- Bork K, Barnstedt SE, Koch P, Traupe H. . Lancet. 2000, 356 (9225): 213–7. PMID 10963200. doi:10.1016/S0140-6736(00)02483-1.
- Cichon S, Martin L, Hennies HC, 等. . Am. J. Hum. Genet. 2006, 79 (6): 1098–104. PMC 1698720. PMID 17186468. doi:10.1086/509899.
- University of Maryland Medical Center. Angioedema. . [2008-01-08]. (原始内容存档于2007-10-12).
- Sabroe RA, Black AK. . British Journal of Dermatology. February 1997, 136 (2): 153–8. PMID 9068723. doi:10.1111/j.1365-2133.1997.tb14887.x.
- Israili ZH, Hall WD. . Annals of Internal Medicine. August 1, 1992, 117 (3): 234–42. PMID 1616218. doi:10.7326/0003-4819-117-3-234.
- Kostis JB, Kim HJ, Rusnak J, Casale T, Kaplan A, Corren J, Levy E. . Archives of Internal Medicine. July 25, 2005, 165 (14): 1637–42. PMID 16043683. doi:10.1001/archinte.165.14.1637.
- Brown NJ, Ray WA, Snowden M, Griffin MR. . Clinical Pharmacologic Therapy. July 1996, 60 (1): 8–13. PMID 8689816. doi:10.1016/S0009-9236(96)90161-7.
- Dykewicz, MS. . Current Opinion in Allergy and Clinical Immunology. August 2004, 4 (4): 267–70. PMID 15238791. doi:10.1097/01.all.0000136759.43571.7f.
- Malde B, Regalado J, Greenberger PA. . Annals of Allergy, Asthma & Immunology. January 2007, 98 (1): 57–63. PMID 17225721. doi:10.1016/S1081-1206(10)60860-5.
- Cicardi M, Zingale LC, Bergamaschini L, Agostoni A. . Archives of Internal Medicine. April 26, 2004, 164 (8): 910–3. PMID 15111379. doi:10.1001/archinte.164.8.910.
- LLC, Prime Therapeutics. . www.prnewswire.com. (原始内容存档于2015-10-25) (英语).
- Drouet C, Désormeaux A, Robillard J, Ponard D, Bouillet L, Martin L, 等. . The Journal of Allergy and Clinical Immunology. 2008, 121 (2): 429–33. PMID 18158172. doi:10.1016/j.jaci.2007.10.048.
- (PDF). [2007-01-26]. (原始内容 (PDF)存档于2007-09-28).
- Quincke H. . Monatsh Prakt Derm. 1882, 1: 129–131.
- synd/482 - Who Named It?
- Marcello Donati. De medica historia mirabili. Mantuae, per Fr. Osanam, 1586
- J. L. Milton. On giant urticaria. Edinburgh Medical Journal, 1876, 22: 513-526.
- Osler W. . Am J Med Sci. 1888, 95 (2): 362–67. doi:10.1097/00000441-188804000-00004. Reprint: PubMed
- Donaldson VH, Evans RR. . Am. J. Med. July 1963, 35: 37–44. PMID 14046003. doi:10.1016/0002-9343(63)90162-1.
- http://healthnews.uc.edu/news/?/24791/ 的存檔,存档日期2014-07-14.
外部链接
| 分類 | |
|---|---|
| 外部資源 |