酶抑制剂
酶抑制剂(英語:)是一类可以与酶结合并降低其活性的分子。 由于阻断酶的活性可以杀死病原体[1]或纠正代谢失衡,许多药物都是酶抑制剂[2][3]。 它们也用于杀虫剂。 并非所有与酶结合的分子都是抑制剂; 酶激活剂与酶结合并增加其酶活性,而酶底物结合并在酶的正常催化循环中转化为产物。

一种抑制剂的结合可以阻止底物进入酶的活性位点和/或阻止酶催化其反应。 抑制剂结合是可逆的或不可逆的。 不可逆抑制剂通常与酶反应并化学改变(例如通过共价键形成)。 这些抑制剂修饰酶活性所需的关键氨基酸残基。 相反,可逆抑制剂非共价结合,并且取决于这些抑制剂是否与酶,和/或酶-底复合物或两者结合,产生不同类型的抑制。
由于抑制特定酶的活性可以杀死病原体或校正新陈代谢的不平衡,许多药物分子都是酶抑制剂,它们的发现和改进是生物化学和药理学研究的一个活跃领域。评判一个药用酶抑制剂通常以它的特异性(不与其他蛋白质结合)及效价(解离常数,表示抑制酶所需的浓度)为指标。高特异性和高效价使药物具有更小的副作用,因而具有更低的毒性。一些酶抑制剂还被用作除草剂或农药。
酶抑制剂天然存在并参与代谢调节。例如,代谢途径中的酶可被下游产物抑制。当产物开始积聚时,这种负反馈会减慢生产的速度,并有助于维持细胞稳态。也有的细胞酶抑制剂是特异性结合并抑制标靶酶的蛋白质。这可以帮助控制可能对细胞有害的酶,如蛋白酶和核酸酶。一个典型例子是核糖核酸酶抑制剂,它可以与核糖核酸酶结合,是已知最紧密的蛋白质-蛋白质相互作用之一。天然酶抑制剂也可以作为毒药,用以防御食肉动物或杀死猎物。
可逆性抑制剂
可逆抑制剂通过非共价相互作用附着于酶,例如氢键,疏水相互作用和离子键。抑制剂和活性位点之间的多个弱键结合产生紧密的特异性结合。与底物和不可逆抑制剂相反,可逆抑制剂在与酶结合时通常不发生化学反应,并且可以通过稀释或透析很容易地除去。
分类
可逆性抑制剂可以根据改变底物浓度对抑制剂的影响分为四种。
竞争性抑制剂
竞争性抑制剂(英語:)在结构上通常与底物相似。它和底物不能同时与酶结合,通常是由于它对酶的活性位点具有亲和力,而底物也与该位点结合,故底物和抑制剂竞争结合酶的活性位点。这种类型的抑制可以通过提高底物浓度,即与抑制剂竞争来克服(此时最大反应速度Vmax保持不变)。然而,表观米氏常数Km(反应速度达到Vmax一半时的底物浓度)将增加,因为需要更高的底物浓度才能达到一半的最大反应速度。[6]
非竞争性抑制剂
非竞争性抑制剂(英語:)通常与酶的非活性部位结合,降低酶的活性,但不影响酶与底物结合。故该抑制剂对反应的抑制程度取决于抑制剂的浓度。由于反应不能有效进行,Vmax将降低,但Km值将保持不变。[6]这是因为Km是酶与底物亲和力的量度,只能通过活性酶来测量。非竞争性抑制中固定量的无活性酶不影响Km,因此不变。[7][8][9]
反竞争性抑制剂
反竞争性抑制剂(英語:)仅与酶-底物复合物结合,导致最大反应速度Vmax因酶-底物复合物被去除而降低。且由于复合物被去除,根据勒夏特列原理可得出酶与底物的结合速率会变快,因此Km值也会降低,酶与底物表现出更高的亲和力。[6]在反应中Vmax和Km的比值均會變小。反竞争性抑制剂在底物浓度高时效果很好。
复合抑制剂
复合抑制剂(英語:)可以与酶的底物同时结合酶。然而,抑制剂的结合影响底物的结合,反之亦然。这种类型的抑制可以被克服,但不能通过增加底物浓度来克服。尽管复合抑制剂可能与酶的活性位点结合,但这种类型的抑制通常是由抑制剂结合酶上的多个位点而产生的变构效应导致。抑制剂与该变构位点的结合改变了酶的构象,从而降低了底物对活性位点的亲和力。
可逆性抑制剂的定量描述
可以根据抑制剂与酶和酶-底物复合物的结合及其对酶的动力学常数的影响来定量描述可逆抑制。在下面的经典米氏动力学模型中,酶(E)与其底物(S)结合以形成酶-底物复合物(ES)。催化后,该复合物分解释放产物(P)和游离酶。抑制剂(I)可以分别以解离常数Ki或Ki′结合E或ES。
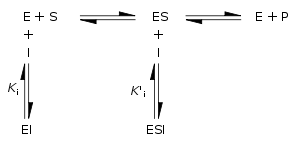
- 竞争性抑制剂可以与E结合,但不与ES结合。竞争性抑制增加Km(抑制剂干扰底物结合),但不影响Vmax(抑制剂不能与ES结合,足够多的底物仍可使反应达到原最大速度)。
- 非竞争性抑制剂对E和ES具有相同的亲和力(Ki = Ki′)。非竞争性抑制不改变Km(抑制剂不影响底物结合),但降低Vmax(抑制剂与ES的结合阻碍催化作用)。
- 反竞争性抑制剂与ES结合,同时降低Km(提高酶与底物亲和力)和Vmax(阻碍催化作用),二者比值保持不变。
- 复合抑制剂与E和ES结合,但它们对二者的亲和力是不同的(Ki ≠ Ki′)。复合抑制剂干扰底物结合(增加Km)并阻碍ES复合物中的催化作用(降低Vmax)。
当酶具有多种底物时,抑制剂可以根据所抑制的底物表现不同类型的抑制。这是由于活性位点内含有不同的结合位点产生的,每个底物对应一个。例如,某抑制剂可能与底物A竞争第一个结合位点,但在第二个结合位点是底物B的非竞争性抑制剂。
解离常数测量
如上所述,酶抑制剂的特征在于对酶和酶-底物复合物的两个解离常数Ki和Ki′。酶抑制剂常数Ki可以通过各种方法直接测量:一种极其准确的方法是等温滴定量热法,这种方法将抑制剂滴定到酶溶液中并测量释放或吸收的热量以测定解离常数。[10]然而,解离常数Ki′难以直接测量,因为酶-底物复合物的存在时间较短,并且易发生生成酶与产物的化学反应。因此,Ki′通常通过观察各种底物和抑制剂浓度下的酶活性,并将数据[11]拟合到稍有改动的米氏动力学方程

中间接测量,其中修饰因子α和α′由抑制剂浓度和它的两个解离常数


来定义。
因此,在抑制剂存在下,酶的有效Km和Vmax分别变为(α/α′)Km和(1/α′)Vmax。然而,改动的米氏动力学方程假定抑制剂与酶的结合已达到平衡,这对于具有亚纳摩尔级别解离常数的抑制剂可能是非常缓慢的过程。在这些情况下,将紧密结合的抑制剂作为不可逆抑制剂通常更为实际(参见下文);然而,如果Ki是独立测量的,那么仍然可以动态地估计Ki′。
可以使用米氏动力学方程的图象表示来显示不同类型的可逆酶抑制剂对酶活性的影响,例如双倒数图和Eadie-Hofstee图象。然而,从这些图中准确估计Ki和Ki′可能很困难[12] ,因此使用非线性回归方法估算这些常数更为可靠,如上所述。
不可逆性抑制剂
不可逆抑制的类型(共价失活)
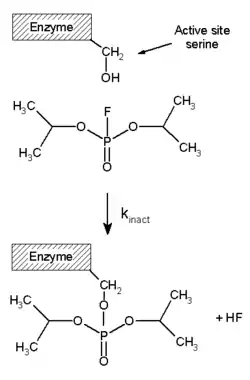
不可逆抑制剂通常通过与酶形成共价键来抑制酶的活性,因此这种抑制不能逆转。不可逆抑制剂通常含有活性官能团,例如氮芥,醛,卤代烷,烯烃,迈克尔受体,苯磺酸盐,MAFP等。这些亲电体与氨基酸侧链反应形成共价加合物,修饰含亲核体(如羟基或巯基)侧链的残基,包括氨基酸丝氨酸(见右侧图),半胱氨酸,苏氨酸,酪氨酸等[13]。
不可逆抑制不同于不可逆的酶失活(enzyme inactivation)。 不可逆抑制剂通常对一类酶具有特异性,不会使所有蛋白质失活; 它们不通过破坏蛋白质结构而是通过特异性改变其靶标的活性位点起作用。 例如,极端的pH或温度通常会导致所有蛋白质结构的变性,但这是非特异性的影响。 同样,一些非特异性的化学处理会破坏蛋白质结构:例如,在浓盐酸中加热会使保持蛋白质的肽键水解,释放游离的氨基酸[14]。
不可逆抑制剂显示出时间依赖性抑制,因此其效力不能通过IC50值表征。 这是因为在给定浓度的不可逆抑制剂下活性酶的量将根据抑制剂与酶预孵育的时间而不同。 相反,kobs/[I]值被使用[15],其中kobs是被观察到的伪一阶失活速率(通过绘制%活性对时间的对数图得到),[I]是抑制剂的浓度。 只要抑制剂不与酶结合(在这种情况下kobs = kinact),kobs/[I]参数就是有效的。
不可逆抑制的分析
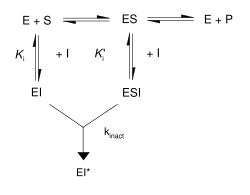
如右图所示,不可逆抑制剂与酶(EI或ESI)形成可逆的非共价复合物,然后反应产生共价修饰的"末端复合物(dead-end complex)"EI*。 EI*形成的速率称为失活速率或kinact。 由于EI的形成可能与ES竞争,因此可通过与底物或第二种可逆抑制剂竞争来预防不可逆抑制剂的结合。 这种保护作用是不可逆抑制剂与活性位点特异性反应的良好证据。
通过将酶与抑制剂一起孵育并测定随时间保留的活性量来研究该反应的结合和失活步骤。 活动将以时间依赖的方式减少,通常跟随指数衰减。 将这些数据拟合至速率方程,给出该抑制剂在这个浓度下的失活速率。 这是在抑制剂的几种不同浓度下完成的。 如果一个可逆的EI复合物被涉及,则失活率将是可饱和的,并且拟合该曲线将给出kinact和Ki</sub [16]。
在这些分析中广泛使用的另一种方法是质谱法。 在这里,准确测量未修饰的天然酶和灭活酶的质量会使与抑制剂反应引起的质量增加,并显示反应的化学计量[17]。 这通常使用基质辅助激光解吸/电离-TOF(MALDI-TOF)质谱仪完成。在一个互补技术中,肽质量指纹识别涉及用蛋白酶如胰蛋白酶消化天然的和修饰的蛋白质。 这将产生可以使用质谱仪分析的一组肽。 与抑制剂反应后质量变化的肽将是含有修饰位点的肽。
发现与设计

新的药物是长期药物研发过程中的产物,而发现新药的第一步往往是发现新的酶抑制剂。在过去,发现新抑制剂的唯一途径是反复实验:筛选大量的化合物对抗靶酶 ,并希望出现一些有效的引物。这种暴力的方法仍然是成功的,甚至通过组合化学方法扩展,快速生产大量的新化合物,并使用高效筛选技术快速筛选这些巨大的化合物库来寻找有效的抑制剂。[18]
最近,一种新方法被广泛应用:合理药物设计利用酶活性位点的三维结构来预测哪些分子可能是抑制剂。[19]对这些预测出的化合物进行测试,新型抑制剂就可能会从这些化合物中产生。然后,对这种抑制剂与酶形成的抑制剂—酶复合物进行测试并获得酶的结构,以显示分子如何与活性位点结合,从而可以对抑制剂进行修改以优化其与酶的结合。重复该测试和改进,直到产生足够有效的抑制剂。[20]使用计算机预测酶抑制剂亲和力的方法也正在开发中,例如分子对接和分子力学。
应用
酶抑制剂存在于自然界中,并且还作为药理学和生物化学的一部分设计和生产。 天然毒物通常是酶抑制剂,其已经进化以保护植物或动物免受捕食者的侵害。 这些天然毒素包括一些已知的毒性最强的化合物。 人工抑制剂通常用作药物,但也可以是杀虫剂,如马拉硫磷,除草剂,如草甘膦,或消毒剂,如三氯生。 其他人工酶抑制剂可阻断乙酰胆碱酯酶,这是一种分解乙酰胆碱的酶,可用作化学战中的神经毒剂。
化学治疗
 西地那非(伟哥)的结构 |
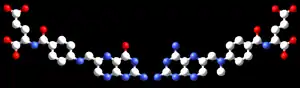 辅酶叶酸(左)与抗癌药物甲氨蝶呤(右)相比较 |
酶抑制剂最常见的用途是作为治疗疾病的药物。 许多这些抑制剂靶向人酶并且旨在纠正病理状况。 然而,并非所有药物都是酶抑制剂。 一些例如抗癫痫药物通过引起或多或少的酶产生来改变酶活性。 这些作用被称为酶诱导和抑制,并且是基因表达的改变,这与本文讨论的酶抑制类型无关。 其他药物与不是酶的细胞靶标相互作用,例如离子通道或膜受体。
药用酶抑制剂的实例是西地那非(伟哥),其是男性勃起功能障碍的常见治疗方法。 该化合物是cGMP特异性磷酸二酯酶5型的有效抑制剂,该酶降解信号分子环磷酸鸟苷[21]。 这种信号分子触发平滑肌松弛,并允许血液流入阴茎海绵体,从而引起勃起。 由于药物降低了停止信号的酶的活性,因此它使该信号持续更长的时间。
另一个实例一些抑制剂与它们靶向的酶底物结构相似性在图中可以被看到,比较药物甲氨蝶呤与叶酸。 叶酸是二氢叶酸还原酶的底物,二氢叶酸还原酶是一种参与制备被甲氨蝶呤有效抑制的核苷酸的酶。 甲氨蝶呤阻断二氢叶酸还原酶的作用,从而停止核苷酸的产生。 这种核苷酸生物合成块对快速生长的细胞比非分裂细胞毒性更大,因为快速生长的细胞必须进行DNA复制,因此甲氨蝶呤经常用于癌症的化学疗法[22]。
抗生素
药物也被用于抑制病原体存活所需的酶[1][2][8][9][3]。 例如,细菌被称为肽聚糖的网状聚合物制成的厚细胞壁包围。 许多抗生素如青霉素和万古霉素可以抑制产生这种聚合物链的酶,然后将这些聚合物交联在一起[23]。 这导致细胞壁失去强度并且细菌破裂。 在该图中,显示青霉素分子(以球-棒形式显示)与其靶标结合,来自细菌链霉菌R61的转肽酶(蛋白质被显示为带状图)。
当对病原体存活至关重要的酶在人体中不存在或非常不同时,促进抗生素药物设计。 在上面的例子中,人类不制备肽聚糖,因此该过程的抑制剂对细菌具有选择性毒性。 通过利用细菌中核糖体结构的差异或它们如何产生脂肪酸,抗生素也产生选择性毒性。
代谢控制
酶抑制剂在代谢控制中也很重要。 细胞中的许多代谢途径被通过变构调节或底物抑制来控制酶活性的代谢物抑制。一个很好的例子是糖酵解途径的变构调节。该异化作用途径消耗葡萄糖并产生ATP,NADH,和丙酮酸。 调节糖酵解的关键步骤是磷酸果糖激酶-1(PFK1)催化的途径中的早期反应。 当ATP水平升高时,ATP结合PFK1中的变构位点以降低酶反应的速率; 糖酵解受到抑制,ATP产生下降。 这种负反馈控制有助于维持细胞中ATP的稳定浓度。 然而,代谢途径不仅仅通过抑制来调节,因为酶活化同样重要。 关于PFK1,果糖2,6-二磷酸和ADP是变构活化剂的代谢物的例子[24]。
农药
许多农药都是酶抑制剂。 乙酰胆碱酯酶(AChE)是从昆虫到人类的动物中发现的酶。 通过其将神经递质乙酰胆碱分解成其成分,乙酸和胆碱的机制,神经细胞功能是必不可少的。 这在神经递质中有点不寻常,因为大多数,包括血清素,多巴胺和去甲肾上腺素,都是从突触间隙吸收而不是切割。 大量的AChE抑制剂用于医药和农业。 可逆的竞争性抑制剂,例如,edrophonium,毒扁豆碱和新斯的明,用于治疗重症肌无力和麻醉。 氨基甲酸酯类农药也是可逆AChE抑制剂的实例。 有机磷农药如马拉硫磷,对硫磷和毒死蜱不可逆地抑制乙酰胆碱酯酶。
除草剂草甘膦是3-磷酸莽草酸1-羧基乙烯基转移酶的抑制剂[25],其他除草剂,如磺酰脲类抑制酶乙酰乳酸合成酶。 这些酶都是植物制备支链氨基酸所必需的。 许多其他酶被除草剂抑制,包括脂质和类胡萝卜素生物合成所需的酶以及光合作用和氧化磷酸化过程[26]。
参考文献
- Srinivasan B, Tonddast-Navaei S, Roy A, Zhou H, Skolnick J. . Medicinal Research Reviews. September 2018. PMID 30192413. doi:10.1002/med.21538.
- Srinivasan B, Tonddast-Navaei S, Skolnick J. . European Journal of Medicinal Chemistry. October 2015, 103: 600–14. PMC 4610388. PMID 26414808. doi:10.1016/j.ejmech.2015.08.021.
- Srinivasan B, Skolnick J. . The FEBS Journal. May 2015, 282 (10): 1922–38. PMC 4445455. PMID 25703118. doi:10.1111/febs.13244.
- (英文)Cleland, W.W. . Biochim. Biophys. Acta. 1963, 67: 173–187.
- (英文)Cleland, W.W. . Biochim. Biophys. Acta. 1963, 67: 173–187.
- .
- (英语).
- Srinivasan B, Rodrigues JV, Tonddast-Navaei S, Shakhnovich E, Skolnick J. . ACS Chemical Biology. July 2017, 12 (7): 1848–1857. PMC 5819740. PMID 28525268. doi:10.1021/acschembio.7b00175 (英语).
- Srinivasan B, Rodrigues JV, Tonddast-Navaei S, Shakhnovich E, Skolnick J. . ACS Chemical Biology. May 2018, 13 (5): 1407. PMID 29688000. doi:10.1021/acschembio.7b00759.
- Holdgate, GA. . BioTechniques. 2001, 31 (1): 164–6, 168, 170 passim. PMID 11464510 (英语).
- Leatherbarrow, RJ. . Trends in Biochemical Sciences. 1990, 15 (12): 455–8. PMID 2077683. doi:10.1016/0968-0004(90)90295-M (英语).
- Tseng, SJ; Hsu, JP. . Journal of Theoretical Biology. 1990, 145 (4): 457–64. PMID 2246896. doi:10.1016/S0022-5193(05)80481-3.
- Lundblad R. L. Chemical Reagents for Protein Modification CRC Press Inc (2004) ISBN 0-8493-1983-8
- Price N, Hames B, Rickwood D. . BIOS Scientific Publishers. 1996. ISBN 978-0-12-564710-6.
- Adam GC, Cravatt BF, Sorensen EJ. . Chemistry & Biology. January 2001, 8 (1): 81–95. PMID 11182321. doi:10.1016/S1074-5521(00)90060-7.
- Maurer T, Fung HL. . AAPS PharmSci. 2000, 2 (1): 68–77. PMC 2751003. PMID 11741224. doi:10.1208/ps020108.
- Loo JA, DeJohn DE, Du P, Stevenson TI, Ogorzalek Loo RR. . Medicinal Research Reviews. July 1999, 19 (4): 307–19. PMID 10398927. doi:10.1002/(SICI)1098-1128(199907)19:4<307::AID-MED4>3.0.CO;2-2.
- Koppitz M, Eis K; Eis. . Drug Discov. Today. 2006, 11 (11–12): 561–8. PMID 16713909. doi:10.1016/j.drudis.2006.04.005.
- Scapin G. . Curr. Pharm. Des. 2006, 12 (17): 2087–97. PMID 16796557. doi:10.2174/138161206777585201.
- Gohlke H, Klebe G; Klebe. . Angew. Chem. Int. Ed. Engl. August 2002, 41 (15): 2644–76. PMID 12203463. doi:10.1002/1521-3773(20020802)41:15<2644::AID-ANIE2644>3.0.CO;2-O.
- Maggi M, Filippi S, Ledda F, Magini A, Forti G. . European Journal of Endocrinology. August 2000, 143 (2): 143–54. PMID 10913932. doi:10.1530/eje.0.1430143.
- McGuire JJ. . Current Pharmaceutical Design. 2003, 9 (31): 2593–613. PMID 14529544. doi:10.2174/1381612033453712.
- Katz AH, Caufield CE. . Current Pharmaceutical Design. 2003, 9 (11): 857–66. PMID 12678870. doi:10.2174/1381612033455305.
- Okar DA, Lange AJ. . BioFactors. 1999, 10 (1): 1–14. PMID 10475585. doi:10.1002/biof.5520100101.
- Tan S, Evans R, Singh B. . Amino Acids. March 2006, 30 (2): 195–204. PMID 16547651. doi:10.1007/s00726-005-0254-1.
- Duke SO. . Environmental Health Perspectives. July 1990, 87: 263–71. JSTOR 3431034. PMC 1567841. PMID 1980104. doi:10.2307/3431034.
外部連結
- Web tutorial on enzyme inhibition, Tutorial by Dr Peter Birch of the University of Paisley, containing very clear animations
- Symbolism and Terminology in Enzyme Kinetics, Recommendations of the Nomenclature Committee of the International Union of Biochemistry (NC-IUB) on enzyme inhibition terminology
- PubChem from NCBI 页面存档备份,存于, Database of drugs and enzyme inhibitors
- BRENDA, Database of enzymes giving lists of known inhibitors for each entry
- Enzymes, Kinetics and Diagnostic Use, On-line lecture concentrating on medical applications of enzyme inhibitors: by Dr. Michael W. King of the IU School of Medicine
- BindingDB, a public database of measured protein-ligand binding affinities.
- Enzyme Inhibition Animated Exercise (tutorial + quizzes).
