维蒂希反应
维蒂希反应(Wittig反应)是醛或酮与三苯基磷鎓內鹽(维蒂希试剂)作用生成烯烃和三苯基氧膦的一类有机化学反应,以发明人德国化学家格奥尔格·维蒂希的姓氏命名。[1][2]

| 维蒂希反应 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 命名根据 | 格奥尔格·维蒂希 | ||||||||||
| 反应类型 | 碳-碳键形成反应 | ||||||||||
| 反应 | |||||||||||
| |||||||||||
| 反应条件 | |||||||||||
| 常用溶剂 | 通常用四氢呋喃或乙醚 | ||||||||||
| 标识 | |||||||||||
| 马奇《高等有机化学》章节 | 16–44(第6版) | ||||||||||
| 有机化学网站对应网页 | wittig-reaction | ||||||||||
| RSC序号 | RXNO:0000015 | ||||||||||
| | |||||||||||
格奥尔格·维蒂希在1954年发现该反应,并因此获得1979年诺贝尔化学奖。[3][4][5] 维蒂希反应在烯烃合成中有十分重要的地位。
维蒂希反应的反应物一般是醛/酮和单取代的磷鎓內鹽。使用活泼叶立德时所得产物一般都是Z型的,或Z/E异构体比例相当;而使用比较稳定的叶立德时,或在Schlosser改进的条件下,产物则以E型为主。
反应机理
经典机理
维蒂希反应的经典机理为:

磷叶立德1中的电负性碳进攻与醛酮羰基2中的碳原子,发生亲核加成。由于位阻原因,主要生成Ph3P+-和-O−处于反式的产物3。3C-C键旋转得到偶极中间体4。4在-78°C时比较稳定。然后生成含氧四元环过渡态5。5发生消除得到顺式烯烃7和三苯基氧膦6。
对于活泼的维蒂希试剂而言,与醛和酮反应时第一步的速率都较快,但第三步成环反应速率较慢,是速控步。但对于稳定的叶立德而言,R1基团可以稳定碳上的负电荷,第一步是速控步。因此总体的成烯反应速率减小,而且生成的烯烃中E型比例较大。这也是不活泼的维蒂希试剂与有位阻的酮反应很慢的缘故。
近期研究
最近的研究表明,以上的机理并不能解释所有的实验数据。目前主要用核磁共振谱来研究活泼维蒂希试剂的反应中间体。但是对于偶极中间体(3a和3b)是否存在及它们之间的相互转换,现在仍有争议。[6] 有证据显示磷叶立德1可以与羰基化合物2发生π²s/π²a [2+2]环加成反应,直接生成含氧的四元环4a和4b,并且产物5的立体化学与叶立德1和2羰基的加成以及4a和4b中间体之间的平衡有关。[7][8][9]布魯斯·瑪麗安諾夫(Bruce Maryanoff)和Reitz研究了它们之间的平衡,将其称为“立体化学移动”(Stereochemical drift)。
多年以来,维蒂希反应的立体选择性一直被认为与产物烯烃的Z/E标记有关,然而有一些反应物却不遵守这样简单的规律。而且锂盐对反应的立体化学似乎也有复杂的影响。[10]

脂肪醛和芳香醛,以及脂肪族和芳香族的鏻盐在发生维蒂希反应时也有显著的不同。Vedejs等人已经表明直链醛在无锂盐存在时没有可逆反应,整个反应是动力学控制的。[11][12] Vedejs也因此提出一套理论来解释活泼和稳定维蒂希反应之间的差别。[13]
维蒂希试剂
简单维蒂希试剂的制备
维蒂希试剂(Wittig)通常以四级鏻盐在强碱作用下失去一分子卤化氢制备,而鏻盐则可由三苯基膦和卤代烃反应得到。前者制备反应通常在乙醚或四氢呋喃中进行,强碱选用苯基锂或正丁基锂。
最简单的维蒂希试剂是亚甲基三苯基膦(Ph3P+−C−H2),是一个橙黄色固体,对空气和水都不稳定,可通过三苯基膦和溴甲烷生成的溴化三苯基甲基鏻 Ph3P+-CH3·Br− 在干燥乙醚和氮气流下用苯基锂处理失溴化氢制得:[14]
- Ph3P + CH3Br → Ph3P+-CH3·Br− -(干燥乙醚,PhLi)→ Ph3P+-CH2−
它也是另一种合成维蒂希试剂方法的原料。合成时一般不将它分离出来,而直接进行下一步的反应。
至于取代叶立德,可先用卤代烃R−CH2−X烷基化Ph3P=CH2,得到一个取代的鏻盐:
- Ph3P=CH2 + R-CH2-X → Ph3P+−CH2− CH2−R X−
再用C4H9Li脱去质子,生成Ph3P=CH−CH2−R。

应用和限制
由于应用性广泛,维蒂希反应已经成为烯烃合成的重要方法。它与消除反应(例如卤代烃的脱卤化氢反应)不同的是,消除反应得到由查依采夫规则决定的结构异构体的混合物,而维蒂希反应得到双键固定的烯烃。
很多醛和酮都可发生该反应,但羧酸衍生物(如酯)反应性不强。因此大多数情况下,单、二和三取代的烯烃都可以较高产率通过该反应制得。羰基化合物可以带着-OH、-OR、芳香-NO2甚至酯基官能团进行反应。
有位阻的酮类反应效果不理想,反应较慢且产率不高,尤其是在与稳定的叶立德反应时。可以用Horner-Wadsworth-Emmons反应来弥补这个不足。而且该反应对不稳定的醛类也不是很适合,包括易氧化、聚合或分解的醛。在“Tandem氧化维蒂希反应”中,维蒂希反应中的醛是由相应的醇在原地氧化获得的。[15]
由于以二级卤代烷作原料生成鏻盐的产率很低,因此维蒂希试剂一般由一级卤代烷反应得到。这意味着四取代的烯烃最好通过其它方法来制取。但维蒂希试剂对很多基团都有很好的耐受性,包括烯烃、芳香环、醚类甚至酯基和与叶立德共轭的C=O和氰基。含两个P=C键的双叶立德也已成功制得并应用于反应中。
此外还有一个与产物立体化学相关的限制。对于简单的叶立德,产物主要是Z型,用酮反应时E型比例高些。而当反应在DMF中和LiI或NaI存在下反应时,产物却几乎全都是Z型的。[16] 这种情况下可以通过Schlosser改进来获得E型产物。对于稳定的叶立德和Horner-Wadsworth-Emmons反应,产物主要为E型。
Schlosser改进

传统维蒂希反应的主要限制在于,反应主要经由赤型的偶极中间体,从而导致Z型烯烃的生成。但Schlosser和Christmann[17] 发现,在低温及苯基锂和HCl的存在下,苏型的中间体占主要地位,因此主要产物为E型烯烃。
科里和山本进一步发现,通过用偶极中间体叶立德与一个二级醛反应,该改进可以被用于烯丙醇的立体选择性合成。[18] 例如:
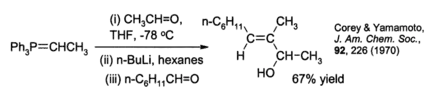
应用举例

由于该反应具有很强的应用性,因此它已经成为有机合成化学家十分重要的工具。[19]
最常见的应用,即是用亚甲基三苯基膦(Ph3P=CH2)向分子中引入亚甲基。在上面的例子中,即便是樟脑这种有位阻的酮,都可通过与甲基三苯基溴化鏻和叔丁醇钾共热(产生维蒂希试剂)而被转化为其亚甲基衍生物。[20] 在另外一个例子中,以氨基钠作为碱产生叶立德,成功以62%的产率将反应物醛转化为烯烃I。[21] 这个反应是低温在THF中进行的,比较敏感的硝基、偶氮基和酚盐负离子都没有干扰反应。产物可用作聚合物的光稳定剂,防止聚合物被紫外线破坏。
白三烯A甲酸酯的合成中也涉及到了维蒂希反应。[22][23] 第一步使用了一个稳定的叶立德,其中羰基与叶立德共轭以防止自身缩合,但还是意外地得到了顺式为主的产物。第二个维蒂希反应则使用的是一个活泼的维蒂希试剂,产物也主要是顺式。需要注意的是环氧化合物和酯都没有对反应造成干扰。

甲氧基亚甲基三苯基膦是一个维蒂希试剂,可用于醛的同系化反应。
参考资料
- Georg Wittig, Ulrich Schöllkopf. . Chemische Berichte. 1954, 87: 1318. doi:10.1002/cber.19540870919.
- Georg Wittig, Werner Haag. . Chemische Berichte. 1955, 88: 1654–1666. doi:10.1002/cber.19550881110.
- Maercker, A. Org. React. 1965, 14, 270-490. (Review)
- W. Carruthers, Some Modern Methods of Organic Synthesis, Cambridge University Press, Cambridge, UK, 1971, pp81-90. (ISBN 0-521-31117-9)
- R. W. Hoffmann. . Angewandte Chemie International Edition. 2001, 40 (8): 1411–1416. doi:10.1002/1521-3773(20010417)40:8%3C1411::AID-ANIE1411%3E3.0.CO;2-U.
- E. Vedejs and C. F. Marth. . J. Am. Chem. Soc. 1990, 112 (10): 3905–3909. doi:10.1021/ja00166a026.
- B. E. Maryanoff, A. B. Reitz, M. S. Mutter, R. R. Inners, and H. R. Almond, Jr., "Detailed Rate Studies on the Wittig Reaction of Non-Stabilized Phosphorus Ylides via 31P, 1H, and 13C NMR Spectroscopy. Insight into Kinetic vs. Thermodynamic Control of Stereochemistry", J. Am. Chem. Soc., 107, 1068-1070 (1985)
- B. E. Maryanoff, A. B. Reitz, D. W. Graden, and H. R. Almond, Jr., "NMR Rate Study on the Wittig Reaction of 2,2-Dimethylpropanal and Tributylbutylidene-phosphorane", Tetrahedron Lett., 30, 1361-1364 (1989)
- B. E. Maryanoff, A. B. Reitz, M. S. Mutter, R. R. Inners, H. R. Almond, Jr., R. R. Whittle, and R. A. Olofson, "Stereochemistry and Mechanism of the Wittig Reaction. Diastereomeric Reaction Intermediates and Analysis of the Reaction Course", J. Am. Chem. Soc., 108, 7664-7678 (1986)
- A. B. Reitz, S. O. Nortey, A. D. Jordan, Jr., M. S. Mutter, and B. E. Maryanoff, "Dramatic Concentration Dependence of Stereochemistry in the Wittig Reaction. Examination of the Lithium-Salt Effect", J. Org. Chem., 51, 3302-3308 (1986)
- E. Vedejs, C. F. Marth and R. Ruggeri. . J. Am. Chem. Soc. 1988, 110 (12): 3940–3948. doi:10.1021/ja00220a036.
- E. Vedejs and C. F. Marth. . J. Am. Chem. Soc. 1988, 110 (12): 3948–3958. doi:10.1021/ja00220a037.
- Vedejs, E.; Peterson, M. J. Top. Stereochem. 1994, 21, 1.
- 邢其毅等。《基础有机化学》第三版上册。北京:高等教育出版社,2005年。ISBN 7-04-016637-2
- Richard J. K. Taylor, Leonie Campbell, and Graeme D. McAllister (2008). "(±) trans-3,3'-(1,2-Cyclopropanediyl)bis-2-(E)-propenoic Acid, Diethyl Ester: Tandem Oxidation Procedure (TOP) using MnO2 Oxidation-Stabilized Phosphorane Trapping 的存檔,存档日期2011-06-05.". Org. Synth. 85: 15-26.
- L. D. Bergelson and M. M. Shemyakin. . Angew. Chem. 1964, 3 (4): 250–260. doi:10.1002/anie.196402501.
- M. Schlosser and K. F. Christmann. . Angewandte Chemie International Edition in English. 1966, 5 (1): 126. doi:10.1002/anie.196601261.
- E. J. Corey and H. Yamamoto. . J. Am. Chem. Soc. 1970, 92 (1): 226–228. doi:10.1021/ja00704a052.
- B. E. Maryanoff and A. B. Reitz. . Chem. Rev. 1989, 89 (4): 863–927. doi:10.1021/cr00094a007.
- Fitjer, L.; Quabeck, U. Synthetic Communications 1985, 15(10), 855-864.
- F. A. Bottino, G. Di Pasquale, A. Pollicino, A. Recca and D. T. Clark. . Macromolecules. 1990, 23 (10): 2662–2666. doi:10.1021/ma00212a011.
- I. Ernest, A. J. Main and R. Menasse. . Tetrahedron Letters. 1982, 23 (2): 167–170. doi:10.1016/S0040-4039(00)86776-3.
- E. J. Corey, D. A. Clark, G. Goto, A. Marfat, C. Mioskowski, B. Samuelsson and S. Hammarstroem. . J. Am. Chem. Soc. 1980, 102 (4): 1436–1439. doi:10.1021/ja00524a045.
外部链接
- 维蒂希反应—Organic Syntheses, Coll. Vol. 10, p. 703 (2004); Vol. 75, p. 153 (1998). (文章)
- 维蒂希反应—Organic Syntheses, Coll. Vol. 5, p. 361 (1973); Vol. 45, p. 33 (1965). (文章)