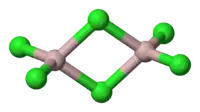
超价分子
超价分子是指由一种或多种主族元素形成,而且中心原子价层电子数超过8的一类分子。例如五氯化磷、六氟化硫、磷酸根离子、三氟化氯以及三碘阴离子都是典型的超价分子。超价分子的概念最早是由上述几种不符合八隅体规则的分子产生的,而自从超价分子的概念提出以来,就处于不断的争议之中。八隅体规则的例外主要有三种,缺电子分子(例如三氟化硼中心原子价电子数为6)、奇电子分子(例如一氧化氮的价电子数是奇数)和超价分子。利用分子轨道理论可以很好地解释前两种分子,然而对于超价分子,不但结构没有得到公认的解释,甚至定义都处于争论之中。
| 未解決的化學問題:超价分子中化学键的本质究竟是什么? |

定义、例子与命名
超价分子的概念最早由杰里米·穆舍尔(Jeremy I. Musher)在1969年正式提出,他定义以15族到18族(包括氮族元素、氧族元素、卤素、稀有气体)元素为中心原子,而且中心原子氧化态比最低氧化态低的分子为超价分子[1]。
例子
以下是一些较为常见的超价分子的例子:
历史
关于超价分子本质和分类方法的争论可追溯到20世纪20年代,即吉爾伯特·路易斯和朗缪尔时期关于化学键本质的争论。路易斯坚持用普通的二中心二电子键(2c-2e)来描述超价分子,从而允许扩大八隅体规则的范围。但另一方面,朗缪尔坚持八隅体规则,并用离子键来解释超价分子,使得价层电子数仍然为8(比如SF42+, F22−)。[3]
20世纪20年代晚期及30年代,萨格登提出二中心一电子键(2c-1e)的存在性,为超价分子的成键提供了无须扩充八隅体规则或引入离子键的解释方法,然而该理论在当时几乎未被接受[3]。20世纪40年代和50年代时,伦德尔和皮门特尔使三中心四电子键理论得到普及,这与萨格登几十年前的理论本质上是相同的。三中心四电子键可被看作两个共线的二中心一电子键组合而成,剩下两个非键电子定域在配体上[3]。
赫尔曼·施陶丁格和格奥尔格·维蒂希在20世纪上半叶进行了制备超价有机分子的尝试,他们寻求挑战当时的化合价理论并成功制备了以氮和磷为中心原子的超价分子[4]。超价的理论基础直到1969年才由穆舍尔基本确立[1]。
1984年,库策尔尼格总结了前人的文献,并用大量确凿的证据证明d轨道参与很少。d轨道参与成键最多只有0.3e,而且主要作用是接受配体反馈的电子,增加体系的稳定性[5]。
1990年,马格努森发表了开创性的成果,明确排除了第2周期元素超价分子中d轨道参与杂化的影响。这是长期以来用分子轨道理论描述这些分子的争论焦点。部份混亂的原因來自於如果使用不含d轨道的基底,就會得出不合理的高能量以及扭曲的分子构型,而且d函数对分子波函数的贡献很大。在历史上這些事實被解讀為d轨道必须参与成键。然而,马格努森总结自己的工作结果后发现,d轨道的参与与超价基本无关[6]。
里德和施莱尔运用6-31G(d)基组,采用哈特里-福克方程和自然布居分析计算了大量超价化合物的键级,表明它们的价层电子数都小于8,符合修正的八隅体规则[7]。
基尔斯洛夫斯基和米克森使用巴德提出的分子中的原子理论(AIM)[8],利用原子重叠矩阵计算超价分子的键级,结果说明它们的离子性很大,中心原子不可能超过8电子[9]。
莫利纳和杜巴多使用电子局域函数(ELF)研究氟化物,结果说明以氟为配体的化合物中心原子价电子数都小于8[10]。
近年来,格莱斯皮[11]和科伯[12]运用多种方法说明超价分子的化学键实际上没有什么特殊之处,所谓修正的八隅体规则是多此一举,大量的量子化学计算反而使得人们难以理解它的成键。然而,格莱斯皮也发现配体与中心原子电负性相近时,电子基本被均分,实际的价电子数依然超过8,例如Te(CH3)6和Se(CH3)6[13]。
争论

超价的名称和概念仍处于争论中。1984年,保罗·冯·拉居·施莱尔(Paul von Ragué Schleyer)根据这项争议提出用超配位这个新名称取代超价,因为新名称不包含对化学键形式的定义,前面的争论因此也可以被回避[3]。
罗纳德·格莱斯皮(Ronald Gillespie)对上述概念提出批评,他以电子定域函数分析为基础,在2002年提出“超价分子和非超价分子(符合八隅体规则)中的化学键没有本质差别,因此没有理由再使用超价这个概念[14]。”
现代量子化学已经证实,对于有高电负性配体的超价分子(例如PF5),高电负性配体可以从中心原子拉走足够的电子云密度,使得中心原子的价层电子数为8甚至更少。关于超价氟化物的事实与这个观点相符,例如与PF5类似的氢化物正膦(PH5)是一种很不稳定的分子。即使是离子键模型也与热化学计算符合得很好。它推测由PF3和F2形成PF4+F−的反应是放热的,有利于自发进行。类似的形成PH4+H−却是吸热的,因此PH5很不稳定[15]。
不符合八隅体规则的分子
引言中已经提到三种不符合八隅体规则的分子,以下简述另外两种。






超价分子的成键
早期科学家鲍林等人研究超价分子的结构,使用人们熟悉的原子成键方式,并运用价层电子对互斥理论解释了一些问题[17][18]。因此,AB5和AB6将分别具有三角双锥和正八面体构型。然而,根据实验所观察到的键角、键长,这种方法明显违反八隅体规则。此外光谱实验表明,d轨道能级较高,成键释放的能量不足以补偿跃迁消耗的能量。例如,四氟化氙5p和5d的能级差高达10电子伏特,这使得sp3dn杂化根本不可能进行[19]。在这以后有几个可供选择的模型被提出[20]。
在20世纪50年代,定域分子轨道理论被引进来解释超价分子的结构。根据这个理论,五配位、六配位的中心原子将分别发生sp3d杂化和sp3d2杂化,这就要求电子跃迁到空的d轨道上。然而,根据量子化学从头计算的结果表明,d轨道对超价分子成键贡献其实很小,因而这种模型不太合理。现在也认为这种杂化轨道理论不太重要[6]。这种方法证明,在六配位的六氟化硫中,d轨道基本没有参与S-F键的形成,但是电荷在硫原子与氟原子之间转移,适当的共振结构可以解释超价分子。
对八隅体规则的补充已经涉及到超价分子成键的离子键特征。作为这些模型的中的一种,三中心四电子键模型在1951年被提出,这个模型使用简单定性的分子轨道来解释超价分子[21]。3c-4e键可以这样描述,中心原子的p轨道与两个配体的轨道线性组合形成分子轨道,这导致被占据的非键轨道成为HOMO,空置的反键轨道成为LUMO。穆舍尔也推荐这种维持八隅体规则的模型[3]。
下面用六氟化硫简单介绍一下该模型的使用,它被认为有三个3c-4e键。在此模型中,每个键都是等价的,分别占据x,y和z轴。这些相互作用是F(p1)-S(3px2)-F(p1), F(p1)-S(3py2)-F(p1)和 F(p1)-S(3pz2)-F(p1)。这种正八面体的结构与实验相符。
超价分子的更完整的描述方法是以量子力学为基础的分子轨道理论,还是用六氟化硫来简单说明[22]。硫的一个3s轨道,三个3p轨道,以及六个八面体构型配位的氟原子对称性匹配的2p轨道进行线性组合,总共得到十个分子轨道(四个全满的成键轨道能量最低,两个全满的非键轨道能量居中,四个全空的反键轨道能量最高),12个价电子填充在上述轨道中。这种稳定的结构只适用于SX6型分子,其中含有高电负性的配体(例如氟原子),这也可以解释为什么SH6不能形成。在硫的成键的模型中,两个非键轨道(1eg)是三中心四电子键的组成部分,使得六个氟原子都是等同的。
结构
五配位磷
对于配体电负性比中心原子更高的超价化合物,可以画出成键电子对不超过四对的共振式,并以离子键与剩余的配体阴离子结合,以符合八隅体规则。例如,五氟化磷通过sp2杂化轨道形成三个水平方向的键。轴向的两个键可以用两种含有一个离子键和一个共价键的共振式来表示,因此它符合八隅体规则,并能解释实验测定的分子构型中轴向与水平方向键长的差异。轴向的键可以表示成两个半键(共振式的对称“平均”结果)或一个三中心四电子键。然而,轴向和水平方向之间的键长的差异大大小于这个结构模型的预测[23]。


六配位磷
六配位的磷化合物分子中含有氮、氧或硫配体,这是路易斯酸-路易斯碱六配位加合物的例子[24]。对于下方画出的两种类似的配合物,随着N-P键的键长变短,C-P键的键长变长;随着N-P路易斯酸碱相互作用的增强,C-P键的键长变短。

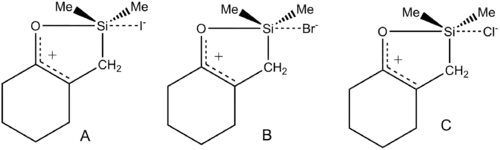

反应
硅
科里于和他的同事对反应特点的早期研究认为四价氯硅烷的水解反应经过了一个超价过渡态[26]。在催化量水存在的情况下测定四价氯硅烷水解反应速率表明,该反应对于氯硅烷是一级反应,而对于水是二级反应。这表明水解过程中两个水分子与硅烷产生相互作用,因此他们提出了双亲核(binucleophilic)反应机理。科里于和他的同事然后测量了亲核催化剂HMPT、DMSO或DMF存在下的水解速率。结果是该反应对于氯硅烷还是一级的,对于催化剂也是一级的,现在对于水也变成了一级反应。水解速率与亲核试剂中氧的亲核性有关也符合设想。
| 氯硅烷 | 亲核催化剂 | 水解速率 (kobs, M-2s-1,20℃) | 氯硅烷 | 亲核催化剂 | 水解速率 (kobs, M-2s-1,20℃) |
|---|---|---|---|---|---|
| Ph3SiCl | HMPT | 1200±100 | MePF2SiCl | HMPT | 2000±200 |
| Ph3SiCl | DMSO | 50±10 | MePF2SiCl | DMSO | 360±50 |
| Ph3SiCl | MDF | 6±1 | MePF2SiCl | DMF | 80±10 |
| Vinyl-1-NpPhSiCl | HMPT | 2200±100 | MePF-1-NpSiCl | HMPT | 3500±400 |
| Vinyl-1-NpPhSiCl | DMSO | 90±10 | MePF-1-NpSiCl | DMSO | 180±20 |
| (m-CF3Ph)-1-NpHSiCl | DMSO | 1800±300 | MePF-1-NpSiCl | DMF | 40±5 |
| (m-CF3Ph)-1-NpHSiCl | DMF | 300±50 |
将两者总结后这个小组提出了反应机理:亲核试剂(或水)首先进攻四配位硅烷形成五配位的超价过渡态,水继续进攻该过渡态形成六配位的活性中间体,迅速分解得到羟基硅烷。
霍姆斯和他的同事对硅烷水解进行了进一步研究[27],四配位的Mes2SiF2(Mes = 苄叉苯基)和五配位的Mes2SiF3-分别与两倍物质的量的水反应。 二十四小时之后,四配位的硅烷几乎没有任何水解迹象,而五配位的硅烷在十五分钟之内就水解完全了。此外,氟硅烷四乙基铵盐的X射线衍射数据中二聚硅醇氢盐的衍射信号支持六配位中间体的存在,此后HF2-被迅速取代得到羟基化的产物。该反应以及晶体结构数据支持科里于等人的想法。

通过格林尼亚反应可知,与类似的四配位化合物相比显然超价分子的反应性更高。科里于小组通过核磁共振测量了相关的18-冠-6钾盐的反应速率,进而获得了在催化量亲核试剂存在下多种四配位和五配位甲基苯基氟硅烷的格林尼亚反应半衰期数据[28]。
| 氟硅烷 | 亲核催化剂 | 半衰期(分钟) | 氟硅烷 | 亲核催化剂 | 半衰期(分钟) |
|---|---|---|---|---|---|
| PHMeSiF2 | i-PrMgBr | 780 | PHMeSiF3- | t-BuMgBr | <3 |
| PHMeSiF3- | i-PrMgBr | <5 | PHMeSiF2 | t-BuMgBr | 360 |
| PH3SiF | i-PrMgBr | 320 | PH3SiF2- | i-PrMgBr | 32 |
尽管半反应的方法是不严密的,反应速率差别的在误差范围内可以绘制出反应的图解,亲核试剂对四配位硅烷的预决速步反应不能完全,始终处于中性四配位物质和五配位阴离子之间的平衡状态。然后两分子格氏试剂进行亲核配位形成六配位过渡态,产生预计的产物。

磷
类似的反应性在其他超价化合物中也已经发现,例如多种磷化合物的六配位过渡态已经被证明。正膦和氧代正膦的水解已经得到了研究[29],说明在水中这是一个二级反应。贝尔斯基等人提出,水的预决速步亲核进攻使得五配位磷化合物和六配位的磷化合物处于平衡之中,然后第二个水分子参与的决速步进行质子转移并开环,得到羟基化的产物。

五配位磷化合物的醇解,例如三甲氧基环磷烯在苯甲醇中醇解,也被假定为经过一个类似的八面体过渡态。与水解不同的是,不经过开环这一步[30]。

从这些实验可以发现,与类似的非超价化合物相比,超价分子的反应活性明显增加。这可以被归结于超价分子的结构与同样超价的过渡态相似,反应过程中所需要的活化能较小。
量子化学从头计算
五配位硅化合物增大的反应性并没有得到完整的解释。科里于和他的同事提出五配位硅原子更高的电正性可能导致了它的反应性增大[31]。初步从头计算在某些程度上支持这个假设,但也使用了一个小的假设作为基础[32]。
迪特尔斯和他的同事使用从头计算的软件程序Gaussian 86来比较四配位的硅或磷化合物与它们的五配位类似物。这种量子化学从头计算方法被用于补充说明五配位化合物的亲核反应活泼性增加的原因。对硅来说,使用6-31+G*基组,因为五配位硅化合物是阴离子;对于磷,则使用6-31G*基组[32]。
理论上五配位化合物比类似的四配位化合物亲电性更弱,因为配体的空间位阻大和电子云密度高,然而实验表明它们的亲核反应活性比四配位的类似物更强。科学家进行了进一步从头计算来更深地理解这类四配位和五配位分子的反应现象。不同系列的物质按氟化程度分类。键长和电子密度的函数可以表示出中心原子连有氢负离子配体的数目。计算时,每增加一个氢原子就减少一个氟原子[32]。
科学家已经通过这种从头计算对于四配位和五配位的硅化合物和磷化合物的键长、电荷密度、马利肯键重叠进行了计算[32]。四配位硅化合物与氟离子的加合总共增加了0.1个元电荷,这被认为是微不足道的。总的来说,三角双锥形五配位化合物中的键长比类似的四配位化合物更长。Si-F键和Si-H键的键长均有所增加,五配位磷化合物的类似现象则较微弱。硅化合物比磷化合物有更显著的键长增加,这是因为配体有效增加了磷的有效核电荷。


此外,迪特尔斯和他的同事[32]发现在所有系列中键长和键重叠程度都是逆相关的。总的来说,五配位的化合物反应活性较高,因为它们的三角双锥结构使得成键较不稳定。
在磷化合物和硅化合物中加合或离去氟离子的能量已经得到了计算。
| 四配位磷化合物 | 焓变(kcal/mol) | 四配位硅化合物 | 焓变(kcal/mol) |
|---|---|---|---|
| -499.7 | -255.7 | ||
| -511.9 | -267.8 | ||
| -528.8 | -282.9 | ||
| -555.8 | -303.1 |








| 五配位磷化合物 | 焓变(kcal/mol) | 五配位磷化合物 | 焓变(kcal/mol) |
|---|---|---|---|
| -199.6 | -33.2 | ||
| -232.7 | -51.3 | ||
| -245.3 | -59.1 | ||
| -245.3 | -66.2 | ||
| -255.8 | -75 |










上面的表格说明,在四配位化合物中离去配体比五配位化合物中需要更高的能量。总的来说,硅化合物中离去配体所需能量比磷化合物低,这是因为硅化合物中的键较弱。
总之,电荷密度改变在计算超配位的硅化合物和磷化合物的反应活性增大时是无意义的。此外,更弱的键造成了反应活性的增加,特别是磷化合物的轴向位置[32]。
应用
这一机理的影响扩展到六配位的硅化合物,这被认为是使用烯丙基三氟硅烷进行醛的烯丙基化反应的过渡态。该反应只在氟离子活化六配位过渡态之前进行,并削弱了六配位过渡态中硅和碳之间的化学键,促使该反应进行[33]。

第二周期超价分子
这类化合物是很罕见的,稳定性也较差由于第2周期元素没有价层d轨道,这使得用杂化轨道理论无法解释它们的成键。但正是它们的不稳定性也说明了d轨道参与成键虽少,但也会使体系的能量下降[34]。
碳

甲鎓离子(CH5+)等碳鎓离子是五配位碳化合物的典型例子,可以认为其中含有一个三中心二电子键[37]。但其中的氢能不断地发生交换反应,甚至绝对零度下也能发生。这使得甲鎓离子成为一种流变分子,核磁共振氢谱上始终只有一个峰[38][39][40]。由于锂的原子半径与氢较接近,还存在CLi5+离子[41]。
双分子亲核取代反应(SN2)的过渡态是一个超价阴离子。一些特殊的阴离子理论上可以较稳定存在,例如[At—C(CN)3—At]-和[Ng—CR3—Ng]-(Ng代表稀有气体原子),它们的空间构型都是三角双锥[42]。
此外,碳与锂形成的化合物CLi5和CLi6相对稳定,它们分解成CLi4和Li2的过程是吸热的[43][44],其他碱金属的类似化合物也有一定稳定性,但不如锂的这类化合物稳定[45]。
碳的超价化合物还不止这些,例如{[(Ph3PAu)6C]2}+[46]、[(Ph3PAu)5C]+[47]和一些更加复杂金原子簇化合物[48]。
氮

从前,科学家认为五氟化氮分子是不可能存在的,离子型的NF4+F-也是不稳定的[49]。然而,理论计算表明,三角双锥型的五氟化氮是三氟化氮与氟分子结合过程中能量最低的状态。根据分子轨道理论,10个电子分别填充在3个强成键轨道(水平方向的键)、1个弱成键轨道(轴向)、1个非键轨道(轴向)中[50]。使用玻恩-哈伯循环计算表明,NF5与NF4F能量仅相差1.0 kJ/mol,都有可能存在。理论上甚至NF6-也是稳定的[51]。
此外,氨还能与铵根离子形成超价聚合物,化学式可表示为NH4(NH3)n(n=1-3)。这类聚合物可由金属钾对[NH4(NH3)n]+的单电子还原制得[52]。
参考文献
- (英文)Musher, J. I. . Angewandte Chemie International Edition in English. 1969-01-01, 8 (1): 54–68. doi:10.1002/anie.196900541.
- (英文)Perkins, C. W.; Martin, J. C.; Arduengo, A. J.; Lau, W.; Alegria, A.; Kochi, J. K. . Journal of the American Chemical Society. 1980-12-01, 102 (26): 7753–7759. doi:10.1021/ja00546a019.
- (英文)Jensen, William B. . Journal of Chemical Education. 2006-12-01, 83 (12): 1751. doi:10.1021/ed083p1751.
- (英文)Akiba, edited by Kin-ya. . New York: Wiley-VCH. 1999. ISBN 0471240192.
- (英文)Kutzelnigg, Werner. . Angewandte Chemie International Edition in English. 1984-04-01, 23 (4): 272–295. doi:10.1002/anie.198402721.
- (英文)Magnusson, Eric. . Journal of the American Chemical Society. 1990-10-01, 112 (22): 7940–7951. doi:10.1021/ja00178a014.
- (英文)Reed, Alan E.; Schleyer, Paul v. R. . Journal of the American Chemical Society. 1990-01-31, 112 (4): 1434–1445. doi:10.1021/ja00160a022.
- (英文)Dobado, J. A.; Martínez-García, Henar; Molina, ; Sundberg, Markku R. . Journal of the American Chemical Society. 1998-08-01, 120 (33): 8461–8471. doi:10.1021/ja980141p.
- (英文)Cioslowski, Jerzy; Mixon, Stacey T. . Inorganic Chemistry. 1993-06-30, 32 (15): 3209–3216. doi:10.1021/ic00067a004.
- (英文)Burdett, Jeremy K.; McCormick, Thomas A. . The Journal of Physical Chemistry A. 1998-07-01, 102 (31): 6366–6372. doi:10.1021/jp9820774.
- (英文)Gillespie, R. . Coordination Chemistry Reviews. 2002-10-31,. 233-234: 53–62. doi:10.1016/S0010-8545(02)00102-9.
- (英文)Cooper, David L. . Theoretical Chemistry Accounts. 2001-04-03, 105 (4-5): 323–327. doi:10.1007/PL00013292.
- (英文)Noury, Stéphane; Silvi, Bernard, Gillespie, Ronald J. . Inorganic Chemistry. 2002-03-31, 41 (8): 2164–2172. doi:10.1021/ic011003v.
- (英文)Gillespie, R. . Coordination Chemistry Reviews. 2002-10-31,. 233-234: 53–62. doi:10.1016/S0010-8545(02)00102-9.
- (英文)Mitchell, Tracy A.; Finocchio, Debbie; Kua, Jeremy. . Journal of Chemical Education. 2007-04-01, 84 (4): 629. doi:10.1021/ed084p629.
- (简体中文)金鑫、李振东. (PDF). 2005. (原始内容 (PDF)存档于2015-06-04).
- (英文)Pauling, Linus. . Journal of Chemical Education. 1992-07-01, 69 (7): 519. doi:10.1021/ed069p519.
- (英文)McWeeny, Roy. 3rd edition. London: Oxford Univ. Pr. 1979. ISBN 978-0198551447.
- (简体中文)《无机化学丛书(第一卷)》,北京:科学出版社,1984
- Xiaoping Sun. . Chem. Educator. 2002, 7: 11–14. doi:10.1007/s00897010525a.
- (英文)Xiaoping Sun. . Chem. Educator. 2002, 7: 261–264. doi:10.1007/s00897020598a.
- (英文)Tachikawa, Hiroto. . Journal of Physics B: Atomic, Molecular and Optical Physics. 2002-01-14, 35 (1): 55–60. doi:10.1088/0953-4075/35/1/304.
- (英文)Gillespie, R.J.; Silvi, B. . Coordination Chemistry Reviews. 2002, 53: 233–234. doi:10.1016/S0010-8545(02)00102-9.
- (英文)Holmes, R.R. . Chem. Rev. 1996, 96: 927–950. PMID 11848776. doi:10.1021/cr950243n.
- (简体中文)张桂玲、戴柏青. . 化学通报. 2000, (10): P46–49.
- (英文)Corriu, RJP. . J. Organomet. Chem. 1978, 150: 27–38. doi:10.1016/S0022-328X(00)85545-X.
- (英文)Johnson, SE; Deiters, JA; Day, RO; Holmes, RR. . J. Am. Chem. Soc. 1989, 111: 3250. doi:10.1021/ja00191a023.
- (英文)Corriu, RJP. . Organometallics. 1988, 7: 237–8. doi:10.1021/om00091a038.
- (英文)Bel'Skii, VE. J. Gen. Chem. USSR. 1979, 49: 298. 缺少或
|title=为空 (帮助) - (英文)Ramirez, F. . J. Am.Chem. Soc. 1968, 90: 751. doi:10.1021/ja01005a035.
- (英文)Corriu, R.; Guerin, C.; Henner, B.; Wong Chi Man, W.W.C. . Organometallics. 1988, 7: 7197–7202. doi:10.1021/ja00176a018.
- (英文)Dieters, J. A.; Holmes, R. R. . J. Am. Chem. Soc. 1990, 112: 7197–7202. doi:10.1021/ja00176a018.
- (英文)Kira, M; Kobayashi, M.; Sakurai, H. . Tetrahedron Letters. 1987, 28: 4081–4084. doi:10.1016/S0040-4039(01)83867-3.
- (英文)Reed, Alan E.; Schleyer, Paul v. R. . Journal of the American Chemical Society. 1990-02-01, 112 (4): 1434–1445. doi:10.1021/ja00160a022.
- (英文)Hirano, Yuichi; Kojima, Satoshi, Yamamoto, Yohsuke. . The Journal of Organic Chemistry. 2011-04-01, 76 (7): 2123–2131. doi:10.1021/jo1024656.
- (英文)Lee, David Y.; Martin, J. C. . Journal of the American Chemical Society. 1984-09-01, 106 (19): 5745–5746. doi:10.1021/ja00331a064.
- (英文)Olah, George A.; Klopman, Gilles; Schlosberg, Richard H. . Journal of the American Chemical Society. 1969-06-01, 91 (12): 3261–3268. doi:10.1021/ja01040a029.
- (英文)Edmund T. White, Jian Tang, Takeshi Oka. . Science. 1999, 284: 135. doi:10.1126/science.284.5411.135.
- (英文)Oskar Asvany, Padma Kumar P, Britta Redlich, Ilka Hegemann, Stephan Schlemmer, Dominik Marx. . Science. 2005, 309 (5738): 1219–22. PMID 15994376. doi:10.1126/science.1113729.
- (英文)Xinchuan Huang, Anne B. McCoy, Joel M. Bowman, Lindsay M. Johnson, Chandra Savage, Feng Dong, David J. Nesbitt. . Science. 2006, 311 (5757): 60–63. PMID 16400143. doi:10.1126/science.1121166.
- (英文)Jemmis, Eluvathingal D.; Chandrasekhar, Jayaraman; Wuerthwein, Ernst Ulrich; Schleyer, Paul von Rague; Chinn, John W.; Landro, Frederick J.; Lagow, Richard J.; Luke, Brian; Pople, John A. . Journal of the American Chemical Society. 1982-07-01, 104 (15): 4275–4276. doi:10.1021/ja00379a051.
- (英文)Pierrefixe, Simon C. A. H.; van Stralen, Sebastiaan J. M., van Stralen, Joost N. P., Fonseca Guerra, Célia, Bickelhaupt, F. Matthias. . Angewandte Chemie International Edition. 2009-08-16, 48 (35): 6469–6471. doi:10.1002/anie.200902125.
- (英文)Kudo, Hiroshi. . Nature. 1992-01-29, 355 (6359): 432–434. doi:10.1038/355432a0.
- (英文)Schleyer, Paul v. R.; Wuerthwein, Ernst Ulrich; Kaufmann, Elmar; Clark, Timothy; Pople, John A. . Journal of the American Chemical Society. 1983-09-01, 105 (18): 5930–5932. doi:10.1021/ja00356a045.
- (英文)Gutsev, G.L.; Boldyrev, A.I. . Chemical Physics Letters. 1982-10-01, 92 (3): 262–266. doi:10.1016/0009-2614(82)80272-8.
- (英文)Scherbaum, Franz; Grohmann, Andreas; Huber, Brigitte; Krüger, Carl; Schmidbaur, Hubert. . Angewandte Chemie International Edition in English. 1988-11-01, 27 (11): 1544–1546. doi:10.1002/anie.198815441.
- (英文)Scherbaum, Franz; Grohmann, Andreas; Müller, Gerhard; Schmidbaur, Hubert. . Angewandte Chemie International Edition in English. 1989-04-01, 28 (4): 463–465. doi:10.1002/anie.198904631.
- (英文)Scherbaum, Franz; Huber, Brigitte; Müller, Gerhard; Schmidbaur, Hubert. . Angewandte Chemie International Edition in English. 1988-11-01, 27 (11): 1542–1544. doi:10.1002/anie.198815421.
- (英文)Grohmann, A.; Riede, J.; Schmidbaur, H. . Nature. 1990-05-09, 345 (6271): 140–142. doi:10.1038/345140a0.
- (英文)Lewars, Errol G. 1. Ed. [Dordrecht]: Springer. 2008. ISBN 978-1-4020-6972-7.
- (英文)Christe, Karl O.; Wilson, William W. . Journal of the American Chemical Society. 1992-11-30, 114 (25): 9934–9936. doi:10.1021/ja00051a027.
- (英文)Gellene, Gregory I.; Porter, Richard F. . The Journal of Physical Chemistry. 1984-12-01, 88 (26): 6680–6684. doi:10.1021/j150670a035.
- (英文)Cahill, Paul A.; Dykstra, Clifford E.; Martin, J. C. . Journal of the American Chemical Society. 1985-10-01, 107 (22): 6359–6362. doi:10.1021/ja00308a032.
